Simulation of Diffusional Association
(version 6)
Additional and auxiliary
programs
SDA-KOFF
This program can use the same input file as sda, doing simulations starting from a docked configuration (as given by pdb files used as input to SDA). The output of this program is a table of residence times (in ps), during which the contacts within a specified distance are maintained. This can be considered to provide an estimate of the lifetime of the complex defined via these contacts.
SDA-SITE
This program calculates the fraction of trajectories starting with one complex (given by the original pdb files and potentials and charges derived from them) and ending with formation of another complex (defined by reaction criteria files). To use it, prepare first standard SDA simulation files using the starting complex of 2 solutes. To define the end-point of trajectories, you can either define reaction criteria manually, or use the SDA routine da-pairs with the same first solute and the second solute docked at the target position. Parameter b (starting sphere) will not be used, parameter c (trajectory truncation distance) is used the same way as in the standard version.
SDA-ENER
This program can use the same input file as sda to read in data about the solute molecules (pdb, charge, grid file names).
It will read in the protein coordinates from the file 'complexes' and write coordinate information and energies to the file 'trajectories' . If the force field parameters in the input file are changed, or another grid/charge files are specified there, then this program will recalculate another set of energies for the same solute configurations. It is important to have the same pdb files for both solutes; charges and grids can be recalculated from these pdb files using different calculation parameters.
SDA3G
This program uses hydrophobic forces derived from a hydrophobic PMF (Lin, Fawzi, Head-Gordon (2007). Structure 15, 727) instead of buried surface area. Use program mk_hd3g_grd.f to generate relevant grids. Usage is the same as the standard SDA program. Note that hdfct in this case is a simple scaling factor to scale the PMF, hdfct=1.0 is the recommended value. Simulations may, however, last a long time with hdfct=1.0 .
ECM
- sda-koff
- sda-site
- sda-ener
- sda3g
- ECM
- mk_LJ_grd
- mk_ed_grd
- mk_hd_grd
- Trajectory Analysis Tool
- other programs in auxi/ subdirectory
SDA-KOFF
This program can use the same input file as sda, doing simulations starting from a docked configuration (as given by pdb files used as input to SDA). The output of this program is a table of residence times (in ps), during which the contacts within a specified distance are maintained. This can be considered to provide an estimate of the lifetime of the complex defined via these contacts.
SDA-SITE
This program calculates the fraction of trajectories starting with one complex (given by the original pdb files and potentials and charges derived from them) and ending with formation of another complex (defined by reaction criteria files). To use it, prepare first standard SDA simulation files using the starting complex of 2 solutes. To define the end-point of trajectories, you can either define reaction criteria manually, or use the SDA routine da-pairs with the same first solute and the second solute docked at the target position. Parameter b (starting sphere) will not be used, parameter c (trajectory truncation distance) is used the same way as in the standard version.
SDA-ENER
This program can use the same input file as sda to read in data about the solute molecules (pdb, charge, grid file names).
It will read in the protein coordinates from the file 'complexes' and write coordinate information and energies to the file 'trajectories' . If the force field parameters in the input file are changed, or another grid/charge files are specified there, then this program will recalculate another set of energies for the same solute configurations. It is important to have the same pdb files for both solutes; charges and grids can be recalculated from these pdb files using different calculation parameters.
SDA3G
This program uses hydrophobic forces derived from a hydrophobic PMF (Lin, Fawzi, Head-Gordon (2007). Structure 15, 727) instead of buried surface area. Use program mk_hd3g_grd.f to generate relevant grids. Usage is the same as the standard SDA program. Note that hdfct in this case is a simple scaling factor to scale the PMF, hdfct=1.0 is the recommended value. Simulations may, however, last a long time with hdfct=1.0 .
ECM
A more detailed description of the ECM program is here .
ECM is located in the directory ecm/ of the SDA distribution.
To run SDA, the effective charges for 2 proteins can be prepared using a script like the one below:
#------------------------------------------
cp p1.pdb dum.pdb
\rm dum.grd; ln -s ep1.grd dum.grd
$ECM_BIN/ecm_mksites < p1.pdb > p1.tcha
$ECM_BIN/ecm_expand < ecm.in > ecm.ou
$ECM_BIN/ecm_regularize >> ecm.ou
$ECM_BIN/ecm_mkreglev 1.0 < ecm.ou > reg_lev
$ECM_BIN/ecm_mkecharges < reg_lev >> ecm.ou
\rm dum.echa dum.echa_E; mv ecm.ou ecm.ou-1
mv dum.echa_R p1.echa
#------------------------------------------
cp p2.pdb dum.pdb; cp p2.tcha dum.tcha
\rm dum.grd; ln -s ep2.grd dum.grd
$ECM_BIN/ecm_mksites < p2.pdb > p2.tcha
$ECM_BIN/ecm_expand < ecm.in > ecm.ou
$ECM_BIN/ecm_regularize >> ecm.ou
$ECM_BIN/ecm_mkreglev 1.0 < ecm.ou > reg_lev
$ECM_BIN/ecm_mkecharges < reg_lev >> ecm.ou
\rm dum.echa dum.echa_E; mv ecm.ou ecm.ou-2
mv dum.echa_R p2.echa
#------------------------------------------
The first part of this script is for the first solute and the second - for the second. To generate effective charges for solute 1, ECM uses its pdb file p1.pdb (in standard PDB file format) and the file with the electrostatic potential ep1.grd (UHBD format). Several ECM routines (located in a directory defined before running the script above as environmental variable ECM_BIN) perform the task of fitting electrostatic potentials to effective charges. The parameter at the command line of the program ecm_mkreglev (1.0 in the script) defines the maximal deviation of the effective charges from the test charge values. The test charge sites and values are computed by ECM routine ecm_mksites and can be modified before executing further steps of the ECM script above.
Input file for the expansion routine can have a form like this :
------------------ pdb file name
dum.pdb
------------------ file with test charges for the molecule
dum.tcha
------------------ grid file name and its UHBD format (0-binary, 1 -ascii)
dum.grd
0
------------------ probe radius (A), skin thickness (A): expansion done in [probe; probe+skin] interval
4.0, 3.0
------------------ ionic strength (mM), solvent relative dielectric constant
50., 78.
------------------ file to write effective charges to
dum.echa
------------------ nothing else
Alternatively, a recent version of ecm does all steps (after assigning test charge sites with ecm_mksites) with a single program, ecm_all, call. No re-calculation of charges using previously calculated fitting results is possible in ecm_all.
Usage:
ecm_all -fp fpdb -ft ftch -fg fgrd -gf gform -pr prob -sk skin -is ios -es epssol -rl reg_lev -rc regch -fe fech
where
| fpdb | pdb file name (dum.pdb) |
| ftch | test charge file name (dum.tcha) |
| fgrd
|
electrostatic potential grid
filename (dum.grd) |
| gform |
ep grid file format, 0 =bin, 1
=ascii (0) |
| prob | probe radius - to construct skin (4Å) |
| skin | skin thickness (3Å) |
| ios | ionic strength in mM (150 mM) |
| epssol | solvent dielectric constant (78.5) |
| reglev | regularisation level |
| regch | max charge deviation after regularisation (1.0 e) |
| fec | effective charge file name (dum.echa) |
mk_LJ_grd
This program is located in the auxi/ directory of the SDA distribution.
mk_LJ_grd reads in parameters from the input file, reads the pdb file for a solute as specified in the input file (p2.pdb in the file below) and writes the Lennard-Jones 12-6 interaction energy grid to the file specified in the input file (apLJ_GRID-00x.grd, x is the number of the site specified in ProbeFile.txt). > mk_LJ_grd < input_file > output_log_file
The input file can look like this:
#------------------------------ h,ndimx,ndimy,ndimz
1.0, 110,110,110
#------------------------------ pfile
p2.pdb
#------------------------------ gfile, iform
LJ_GRID
0
#------------------------------ ProbeFile
ProbeFile.txt
0
#------------------------------ cutoff:
100
Here, the first 4 parameters give the Lennard-Jones grid spacing (Å) and the grid dimensions along the x,y,z directions. The grid will be centered on the molecule specified in pfile. The iform parameter is to define if the UHBD format output file is to be written in binary (0) or ascii (1) format. > The ProbeFile is calculated by the script PrepareProbeFile.py (description in the table below) and describes the atoms whose Lennard-Jones interaction with the molecule specified in pfile is calculated.
The output grid contains the Lennard-Jones interaction energy between the probefile atoms and the pfile solute molecule.
This program is located in the auxi/ directory of the SDA distribution.
mk_LJ_grd reads in parameters from the input file, reads the pdb file for a solute as specified in the input file (p2.pdb in the file below) and writes the Lennard-Jones 12-6 interaction energy grid to the file specified in the input file (apLJ_GRID-00x.grd, x is the number of the site specified in ProbeFile.txt). > mk_LJ_grd < input_file > output_log_file
The input file can look like this:
#------------------------------ h,ndimx,ndimy,ndimz
1.0, 110,110,110
#------------------------------ pfile
p2.pdb
#------------------------------ gfile, iform
LJ_GRID
0
#------------------------------ ProbeFile
ProbeFile.txt
0
#------------------------------ cutoff:
100
Here, the first 4 parameters give the Lennard-Jones grid spacing (Å) and the grid dimensions along the x,y,z directions. The grid will be centered on the molecule specified in pfile. The iform parameter is to define if the UHBD format output file is to be written in binary (0) or ascii (1) format. > The ProbeFile is calculated by the script PrepareProbeFile.py (description in the table below) and describes the atoms whose Lennard-Jones interaction with the molecule specified in pfile is calculated.
The output grid contains the Lennard-Jones interaction energy between the probefile atoms and the pfile solute molecule.
mk_ed_grd
This program is located in the auxi/ directory of the SDA distribution.
mk_ed_grd reads in parameters from the input file, reads the pdb file for a solute as specified in the input file (p1.pdb in the file below) and write the electrostatic desolvation penalty potentials to the file specified in the input file (p1ed.grd). > mk_ed_grd < input_file > output_log_file
The input file can look like this:
#------------------------------ h,ndimx,ndimy,ndimz
1.0, 110,110,110
#------------------------------ iostr,epssol,rion
50. 78. 2.0
#------------------------------ pfile
p1.pdb
#------------------------------ gfile, iform
p1ed.grd
0
Here, the first 4 parameters give the electrostatic desolvation grid spacing (Å) and the grid dimensions along the x,y,z directions. The second set of parameters are for the ionic strength (mM), solvent dielectric constant and ion radius (A) relevant to defining the ionic strength conditions. The last parameter is to define if the UHBD format output file is to be written in binary (0) or ascii (1) format. >
The output grid contains the electrostatic desolvation penalties for a unit charge due to the low dielectric cavity of the solute treated as a collection of atom-centered spheres having radii equal to atomic van der Waals radii, see reference. For a fractional charge, one has to multiply the values on the grid with the square of the charge. For more than one charge, this approach gives approximate desolvation penalties, because no mutual influence of these charges are taken into account . The solute's low dielectric cavity as a region inside a molecular surface can be modeled by using a correction factor to this penalty, see reference .
mk_hd_grd
This program is located in the auxi/ directory of the SDA distribution.
mk_hd_grd reads in parameters from the input file, reads the pdb file for a solute as specified in the input file (p1.pdb in the file below) and write the hydrophobic desolvation penalty potentials to the file specified in the input file (p1hd.grd). Usage:
mk_hd_grd < input_file > output_log_file
The input file can look like this:
#------------------------------ h,ndimx,ndimy,ndimz
1.0, 110,110,110
#------------------------------ a,b,factor
3.10, 4.35, 0.5
#------------------------------ pfile
p1.pdb
#------------------------------ gfile, iform
p1hd.grd
0
Here the first 4 parameters give the hydrophobic desolvation grid spacing (Å) and the grid dimensions along the x,y,z directions. The second set of parameters specifies the region where this potential is defined and the scaling factor for the calculated potential. The last parameter is to define if UHBD format output file is to be written in binary (0) or ascii (1) format.
This program assigns a value of gamma (parameter "factor" in the input file above) to a point if this point is within distance a (in Å) from the surface of the protein, zero if a point is further than b (in Å) from the surface and a linearly interpolated value if a point is inbetween:
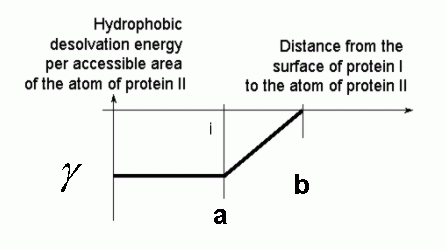
Recommended values are a=3.10 Å
and b=
4.35 Å and factor=0.5.
The value factor=0.5 is to compensate for the factor of 2 in
calculating the buried area by multiplying the solvent accessible areas
of
atoms with the grid values calculated using the above values of a and b. With these parameters, the
procedure used in SDA
gives the most accurate approximation to the buried area. With
this parameters in mk_hd_grd, the parameter hdfct
is a parameter to multiply
the buried area to get the hydrophobic desolvation energy in units
of kcal/mole/Å2. >
If other values of a and b are used, it is recommended to calculate the
hydrophobic desolvation energies for a set of encounter complexes using
SDA and compare them to energies calculated differently to relate SDA
energies to energies calculated using buried areas.
Trajectory Analysis Tool
The trajectory analysis tool including the documentation can be found in auxi/traj_analysis.
Other programs in the auxi/ directory of the SDA distribution.
[Back to Index]
This program is located in the auxi/ directory of the SDA distribution.
mk_ed_grd reads in parameters from the input file, reads the pdb file for a solute as specified in the input file (p1.pdb in the file below) and write the electrostatic desolvation penalty potentials to the file specified in the input file (p1ed.grd). > mk_ed_grd < input_file > output_log_file
The input file can look like this:
#------------------------------ h,ndimx,ndimy,ndimz
1.0, 110,110,110
#------------------------------ iostr,epssol,rion
50. 78. 2.0
#------------------------------ pfile
p1.pdb
#------------------------------ gfile, iform
p1ed.grd
0
Here, the first 4 parameters give the electrostatic desolvation grid spacing (Å) and the grid dimensions along the x,y,z directions. The second set of parameters are for the ionic strength (mM), solvent dielectric constant and ion radius (A) relevant to defining the ionic strength conditions. The last parameter is to define if the UHBD format output file is to be written in binary (0) or ascii (1) format. >
The output grid contains the electrostatic desolvation penalties for a unit charge due to the low dielectric cavity of the solute treated as a collection of atom-centered spheres having radii equal to atomic van der Waals radii, see reference. For a fractional charge, one has to multiply the values on the grid with the square of the charge. For more than one charge, this approach gives approximate desolvation penalties, because no mutual influence of these charges are taken into account . The solute's low dielectric cavity as a region inside a molecular surface can be modeled by using a correction factor to this penalty, see reference .
mk_hd_grd
This program is located in the auxi/ directory of the SDA distribution.
mk_hd_grd reads in parameters from the input file, reads the pdb file for a solute as specified in the input file (p1.pdb in the file below) and write the hydrophobic desolvation penalty potentials to the file specified in the input file (p1hd.grd). Usage:
mk_hd_grd < input_file > output_log_file
The input file can look like this:
#------------------------------ h,ndimx,ndimy,ndimz
1.0, 110,110,110
#------------------------------ a,b,factor
3.10, 4.35, 0.5
#------------------------------ pfile
p1.pdb
#------------------------------ gfile, iform
p1hd.grd
0
Here the first 4 parameters give the hydrophobic desolvation grid spacing (Å) and the grid dimensions along the x,y,z directions. The second set of parameters specifies the region where this potential is defined and the scaling factor for the calculated potential. The last parameter is to define if UHBD format output file is to be written in binary (0) or ascii (1) format.
This program assigns a value of gamma (parameter "factor" in the input file above) to a point if this point is within distance a (in Å) from the surface of the protein, zero if a point is further than b (in Å) from the surface and a linearly interpolated value if a point is inbetween:
In SDA, the solvent accessibilities of
the atoms of the second solute are multiplied by the value of this
grid at
the location of the atoms of the second solute. It appears that many
different values of
parameters a and b give the buried areas to an
accuracy of ca 10%:
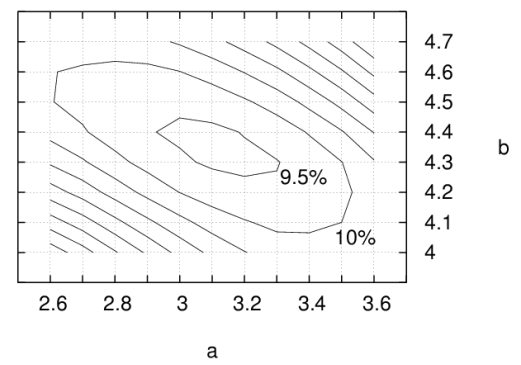
The errors above are for estimating the buried area for a collection of
encounter complexes up to a
constant factor, different values of a and
b approximate the buried area with different factors.
Trajectory Analysis Tool
The trajectory analysis tool including the documentation can be found in auxi/traj_analysis.
Other programs in the auxi/ directory of the SDA distribution.
| apbs2uhbd.f | Convert an APBS potential grid
(exported as
uhbd, in ascii format, in kT/e units) to UHBD format (binary, in
kcal/mole). Default conversion factor is 0.6, the user can specify
another factor on the command line as the third argument. Usage: apbs2uhbd ascii_APBS_file bin_UHBD_file factor_optonal(0.6) |
| ascii2bin.f |
Converts UHBD ascii grid to
binary format. Usage: ascii2bin ascii_file binary_file |
| bin2ascii.f
|
Converts UHBD binary grid to
ascii format. Usage: bin2ascii binary_file ascii_file |
| echa4toecha5.f | Converts effective charges used
in version 4 of SDA to
effective charges used in SDA version 5, see details here. The former charges
are
obtained from the latter by dividing by
the solvent dielectric constant. This is read from the ECM written
REMARK
line of the effective charge file, or set to 78.5 when no such line is
found. If a
switch -inv is given in the command line, this program will do inverse
conversion. Usage: echa4toecha5 < sda4_eff_cha_file > sda5_eff_cha_file echa4toecha5 -inv < sda5_eff_cha_file > sda4_eff_cha_file |
| mk_hd3g_grd.f | Makes a grid of hydrophobic PMF (Lin,
Fawzi, Head-Gordon (2007). Structure 15,
727). These potentials should be used with sda version sda3g
. This program is used the same way as mk_hd_grd, with the input
file of the same form, except that parameters a,b and factor are
not used (although the numbers and lines should be present). |
| ncmx2rmsd_appr.f | Calculates (approximately) the
RMSD
of complexes written in the 'complexes' file to the original (as given in
input pdb
files) complex. Usage: ncmx2rmsd_appr ncoa(optional) < 'complexes' where the optional parameter ncoa forces the program to process only every ncoa-th record in the SDA trajectory file 'complexes'. |
| nda-pairs.f | Calculates donor-acceptor pairs
for a complex given as 2 pdb files. Usage: nda-pairs pdb1f pdb2f cutmax where pdb1f is the filename of the first solute in the complex and pdb2f is the filename for the second solute. cutmax is a maximal distance (in A) between donor and acceptor atoms, default value of 4.5 A is used if this parameter is not given. The results are written to files 1.rxna and 2.rxna - these are donor or acceptor atoms of the first and second solute, respectively; and the atom on the N-th line of 1.rxna is a pair for the atom on the N-th line of 2.rxna. |
| nhis2dcd.f | Converts the trajectory written
by SDA in a '' file to a DCD format. >
Usage: nhis2dcd pdbf hisf outf pdbf - pdb filename of the 2nd (mobile) solute hisf - trajectory file ('trajectories') name outf - output DCD filename |
| nhis2his.f | Rewrites a trajectory file to
another trajectory file with configurations written only every given
time interval (in picoseconds). Usage: nhis2his dt < hisf1 > hisf2 dt - time interval (ps) between snapshots in a new trajectory file hisf1 - original trajectory file hisf2 - new (converted) trajectory file |
| nhis2hisn.f | Rewrites a trajectory file to
another trajectory file with configurations written only every given
number of BD steps. Usage: nhis2his ncoa < hisf1 > hisf2 ncoa - interval between snapshots in a new trajectory file hisf1 - original trajectory file hisf2 - new (converted) trajectory file |
| nhis2pdbs.f | Converts complexes in a file
'complexes' to a set of PDB files. Usage: nhis2pdbs hisf pdbf X nmax hisf - trajectory file pdbf - pdb file for the mobile solute X - first letter for generated pdb files (these file will have names like X1234.pdb) nmax - a number of trajectory records to process |
| nos2rates.f | Extracts rates and
calculates averages and deviations from a set of sda
output files. Usage: nos2rates outf1 outf2 outf3 ... outf1 etc - sda output files |
| nos2times.f |
Extracts residence times
and calculate averages and deviations from a set of sda-koff
output files. Usage: nos2times outf1 outf2 outf3 ... outf1 etc - sda-koff output files |
| nos2fractions.f | Extracts a fraction of reactive
trajectories
and calculates average and deviation from a set of sda-sites
output files. Usage: nos2fractions outf1 outf2 outf3 ... outf1 etc - sda-sites output files |
| prepLJgrids.py | Python script to create the Lennard-Jones interation energy grids for a
given solute molecule and probe. First, it calculates the ProbeFile.txt for a given protein and surface using the
script PrepareProbeFile.py. Then it uses the ProbeFile.txt to calculate the LJ grids with the
program mk_LJ_grd. Usage: ./prepLJgrids.py InputFile [--silent] The --silent parameter is used to run the script in the background. The script overwrites existing files without asking. |
| PrepareProbeFile.py | Python script used for calculating the Lennard-Jones interaction energy between a protein and a surface. The interaction energy is calculated between all atoms of the protein and all atoms of the surface. Usage: ./PrepareProbeFile.py SurfaceFile.pdb ProteinFile.pdb ParameterFile OutputName.txt SurfaceFile - surface structure file in PDB format ProteinFile - solute structure file in PDB format ParameterFile - database with information about charges and radii of common atoms (usually qtable) OutputName.txt - outputfile in txt format. Used as input in mk_LJ_grd. |
| rewrite-pdb.f |
Rewrites ATOM records of pdb
file to locate the first atom name letter at position 13.
Optionally writes only heavy atoms. > Usage: rewrite-pdb < pdb-file > rewritten-pdb-file rewrite-pdb -noh < pdb-file > pdb-file-without-hydrogens |
| DelRotZ_complexes.py |
Python script used to process results of docking a solute to a surface.
It translates the solute coordinates written in 'complexes' parallel to the surface to
x=0,y=0 and saves the new coordinates in fort_wtrotz.fort55. It uses the library, fort55_lib.py. >Usage: DelRotZ_complexes.py 'complexes'-file number-of-records 'complexes'-file - 'complexes' output file from docking simulations number-of-records - number of configurations used for clustering (each line of the 'complexes' file corresponds to one configuration) |
| myClustering_complexes.py |
Python script for clustering docking configurations, uses the output file from
DelRotZ_complexes.py >Usage: My_Clusteryng_f55.py fort_wtrotz.fort55 p2.pdb number-of-records fort_wtrotz.fort55 p2.pdb -output file of DelRotZ_complexes.py p2.pdb - surface structure file in PDB format number-of-records - number of configurations used for clustering |
| generateComplexesPdbs.py |
Python script generating pdb structures of docking confgurations, uses 'complexes' or output file of My_Clusteryng_f55.py >Usage: generateComplexesPdbs.py 'complexes' p2.pdb number-of-configurations name-letter number-of-configurations -number of pdb files to be generated (each file corresponds to one line of 'complexes') p2.pdb - surface structure file in PDB format name-letter - first letter in the names of pdb files |
| rxna2rxnaC.f |
The script converts the two *.rxna files from the SDA5 program to the new format used in SDA6. Run the program with the two filenames as parameter arguments: ./rxna2rxnaC p1.rxna p2.rxna [outFileName] [dist] [specific/nonspecific] outFileName: name of the output file. Default:p12c.rxna dist: constraint distance in Angstroms, any format. Default:6.00 specific/nonspecific: specifies if the constraint is specific (=0) or nonspecific (=1). Default:1 Usage example: ./rxna2rxnaC p1.rxna p2.rxna p12.rxna 6.00 0 |
| convert_sda5_to_sda6_input.f |
Program to convert SDA5 parameter input files to the format used in SDA6.
In addition the program to convert the reaction center files (rxna2rxnaC.f)
is called.
(This depends on the correct definition of the environment variable SDA_DISTR_DIR.
The command is printed out, such that the user
can call the conversion of the reaction centers manually if the environment variable is not defined). Usage example: ./convert_sda5_to_sda6_input new_reaction_center_file < sda5.in > sda6.in |
| change_atomNames.sh |
Script to change hydrogen names from the AMBER (used in *.pqr file) to the OPLS format (used in qtable). Note, that the script overrides the input file! Usage example: ./change_atomNames.sh p2.pdb |
| polar_H_only.py |
Python script to remove non-polar hydrogen atoms from the pdb file Usage example: ./H-away.py p2-H.pdb p2.pdb |
| clust |
Program to cluster docking results. For description see clustering page |
[Back to Index]