Simulation of Diffusional Association
(version 5.01)
Input file
The input file consists of a series of blocks of input parameters. The format of each block of the input file is as follows:
A one (!) line description of a block of input parameters, starting with "#". On the next line, comma or space separated numerical values are given in the required order. Then, the file names (if any) are given in the required order, each on separate lines. The file looks like this (follow the links for detailed descriptions):
# dseed = random_number_generator_seed
256.d0
# nrun, nprint, nrec = no_of_runs, printout_every_nprint_run, record_this_run (or all runs in case of a negative nunber)
100, 5, 0
# p1f, p2f = pdb_filenames_for_solute/protein_1, 2
p1.pdb
p2.pdb
# probe, probew, hexcl, thres - probe_pr2(Å), probe_water(Å), excl_grid_spacing(Å), acc_thres(Å2)
1.70, 1.4, 0.5, 0.0
# dm, dr, dr1, irot2f = tr_diffco, rot_diffco_2, rot_diffco_1, if_prot_1_rotates
0.027, 3.92e-5, 3.92e-5, 1
# dt1, swd1, dt2, swd2, rswd = tstep, tstep_sw_dist_1, tstep_2, tstep_sw_dist_2, rot_sw_dist
0.50, 50.0, 20.0, 90.0, 90.0
# b, c, rboost, novers, djump = b, c, boost_val, no_overs_bef_boost, jump_thr_to_rec
100.0, 500.0, 1.0, 150, 5.0
# icommrxn, dind = reaction_mon_switch, force_switch, indep_dist
3, 6.0
# win0, dwin, nwin, iwrec, nwrec, ncec, rxna1f, rxna2f = rxn_defs, rxn_at_files_prot_1, 2
1.0, 1.0, 50, 1, 40, 500
p1.rxna
p2.rxna
# epfct, ep1f, ep2f, qef1f, qef2f = e-pot_factor, e-pot_for_prot_1, 2, charges_for_prot_1, 2
1.00
ep1.grd
ep2.grd
p1.echa
p2.echa
# edfct, ed1f, ed2f = e-deso_factor, e-deso_file_for_prot_1, 2
1.00
ed1.grd
ed2.grd
# hdfct, hd1f, hd2f = h-deso_factor, skin, h-deso_file_for_prot_1,2
1.00
hd1.grd
hd2.grd
# fnintr = intermediate results filename
sda.int
Detailed description of input parameters
# BLOCK 01: dseed
# BLOCK 07: b,
c, rboost,
novers, djump
# BLOCK 08: icommrxn,
dind
icommrxn=2 - minimal pairwise distance
between pair of atoms given in files rxna1f and
rxna2f, simultaneous occurrence is monitored for any 1, 2, 3 or 4 pairs.
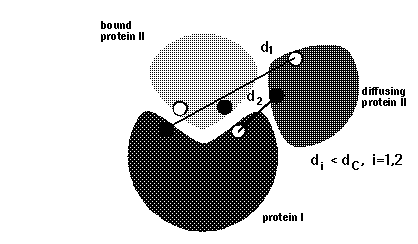
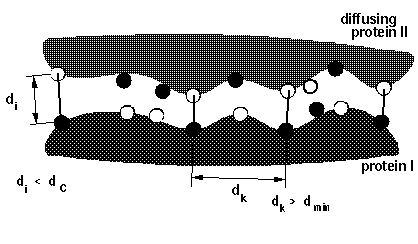
icommrxn=3 - minimal pairwise distance
between a pair of atoms given in files rxna1f and
rxna2f, for independent pairs (defined by dind), simultaneous occurrence is
monitored for any 1, 2, 3 or 4 pairs.
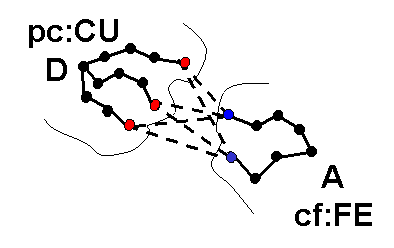
icommrxn=4 - electron transfer rate constant
will be
calculated. Files rxna1f and
rxna2f (defining the reaction atom pairs) should contain pairs of atoms
that may take part in an electron
transfer path together with their coupling to the donor or acceptor
site. In the image below, each possible pairing
(broken line) between surface atoms of proteins 1 (blue) and 2 (red)
should be defined in files rxna1f and
rxna2f.
# BLOCK 09: win0, dwin, nwin, iwrec, nwrec, ncec, rxna1f, rxna2f
# BLOCK 10: epfct, ep1f, ep2f, qef1f, qef2f
The input file consists of a series of blocks of input parameters. The format of each block of the input file is as follows:
A one (!) line description of a block of input parameters, starting with "#". On the next line, comma or space separated numerical values are given in the required order. Then, the file names (if any) are given in the required order, each on separate lines. The file looks like this (follow the links for detailed descriptions):
# dseed = random_number_generator_seed
256.d0
# nrun, nprint, nrec = no_of_runs, printout_every_nprint_run, record_this_run (or all runs in case of a negative nunber)
100, 5, 0
# p1f, p2f = pdb_filenames_for_solute/protein_1, 2
p1.pdb
p2.pdb
# probe, probew, hexcl, thres - probe_pr2(Å), probe_water(Å), excl_grid_spacing(Å), acc_thres(Å2)
1.70, 1.4, 0.5, 0.0
# dm, dr, dr1, irot2f = tr_diffco, rot_diffco_2, rot_diffco_1, if_prot_1_rotates
0.027, 3.92e-5, 3.92e-5, 1
# dt1, swd1, dt2, swd2, rswd = tstep, tstep_sw_dist_1, tstep_2, tstep_sw_dist_2, rot_sw_dist
0.50, 50.0, 20.0, 90.0, 90.0
# b, c, rboost, novers, djump = b, c, boost_val, no_overs_bef_boost, jump_thr_to_rec
100.0, 500.0, 1.0, 150, 5.0
# icommrxn, dind = reaction_mon_switch, force_switch, indep_dist
3, 6.0
# win0, dwin, nwin, iwrec, nwrec, ncec, rxna1f, rxna2f = rxn_defs, rxn_at_files_prot_1, 2
1.0, 1.0, 50, 1, 40, 500
p1.rxna
p2.rxna
# epfct, ep1f, ep2f, qef1f, qef2f = e-pot_factor, e-pot_for_prot_1, 2, charges_for_prot_1, 2
1.00
ep1.grd
ep2.grd
p1.echa
p2.echa
# edfct, ed1f, ed2f = e-deso_factor, e-deso_file_for_prot_1, 2
1.00
ed1.grd
ed2.grd
# hdfct, hd1f, hd2f = h-deso_factor, skin, h-deso_file_for_prot_1,2
1.00
hd1.grd
hd2.grd
# fnintr = intermediate results filename
sda.int
Detailed description of input parameters
# BLOCK 01: dseed
- dseed = random number
generator seed - defines how the
simulations start - different numbers will
result in different sets of trajectories. This parameter can be
given as the first command line parameter to sda - then the value from
the command line will be used.
- nrun = number of runs (trajectories). The higher this number the higher is the number of reactive trajectories, nrun_reactive. The relative error of the calculated rate constant is ~ 1/sqrt(nrun_reactive). This means that to have the same relative error, a larger value of nrun is required in cases with lower association rate constants. This parameter can be given as the second command line parameter to sda - then the value from the command line will be used.
- nprint = frequency with which rate information is printed: the fraction of reactive trajectories is printed every nprint runs. This is most useful when the simulation takes a long time, then intermediate results are available after each nprint trajectories. >
- nrec = number of the
trajectory
to be recorded (nrec > nrun or
nrec equals 0 means no recording). If one wants to record the
reactive trajectory, it is useful to first run SDA with nprint=1, identify
a trajectory in which the number of reactive trajectories changes, and
re-run SDA with nrec set to the number of that trajectory. To record all trajectories,
set this value
to -1. This will use a lot of space.
- p1f = pdb filename for
solute 1. Standard pdb format is required and atomic radii are
assigned
according to the character at position 13 or 14 (if the character at
position 13 is
a space). The atomic radius assignment routine is in van.f - it can be modified to assign different
radii.
- p2f = pdb filename for solute 2
- probe = radius of the
probe representing the surface atoms of the solutes. Namely,
proteins are represented as a collection of
atoms with solvent accessibility larger than thres and each of these atoms are
assigned a
radius of probe (Å),
typical values of this parameter are 1.4-1.9 Å.
- probew = radius of
solvent probe used to calculate the accessibilities of
the solute 2's atoms. A recommended value to use is 1.4 Å. This
value
is especially important if the hydrophobic desolvation term is used,
because the calculated accessibilities are used to calculate
the hydrophobic desolvation energy and forces.
- hexcl = spacing of the
exclusion grid with which a solute
is represented. hexcl
defines
the accuracy with which the shape of the solute is described.
Typical values are 0.5-1.0 Å
for all-atom models of proteins. It needs to be consistent
with the timestep used for BD moves - the spatial accuracy of the
protein representation
should be comparable to an average BD move in realistic simulations.
- thres = accessibility
threshold - only the atoms of solutes
with
accessibility larger than thres
(Å) are used as surface atoms. Moreover, only these atoms are used in
calculating hydrophobic interactions.
- dm = relative translational diffusion coefficient, in Å2/ps. It equals the sum of translational diffusion constants of the two simulated solutes, when both of them are free to move, and equals the translational constant of a mobile (2) solute, if the first solute is fixed. For example, if the association of 2 lysozyme solutes in pure water is simulated, then dm should be 0.022 Å2/ps, because a value of the translational diffusion constant for lysozyme in water is 0.011 Å2/ps. According to the Einstein-Stokes formula, the translational diffusion constant scales inversely proportionally to the solvent viscosity (which itself has a complicated dependence on the temperature), and inversely proportional to the (hydrodynamic) radius of a solute. 1 Å2/ps = 10-8 m2/s = 10-4 cm2/s
- dr = rotational diffusion
coefficient for solute 2 in radian2/ps. Inversely
proportional to the square of the solute radius.
dr
is inversely proportional to the solvent viscosisity
For lysozyme,
dr=2.6*10-5 1/ps.
- dr1 = rotational
diffusion coefficient of solute 1. If irot2f=0, then this
parameter is not used.
- irot2f = switch to define whether the first solute rotates (0 means no rotation and dr1 is not used).
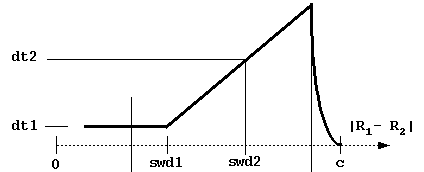
- dt1 = basal simulation
timestep (in ps, picoseconds). This is the smallest timestep used
and is employed when
the center-to-center separation is less than swd1 ori the distance at which
the solutes have the possibility
to contact (i.e. the center-to-center
distance between solutes is less than rhit = the sum of maximal
extents + probe radius + maximal radius of solute 1 atoms).
- swd1 = center-to-center
distance (Å) at which the timestep starts
to increase.
- dt2 = timestep at distance swd2.
- swd2 = center-to-center
distance at which timestep equals
dt2. These parameters define the variable timestep designed to
accelerate simulations.
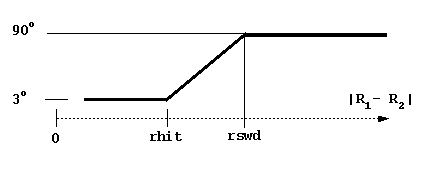
- rswd = rotation switch
distance (see figure below, rhit here is again the
distance at
which the solutes have a the possibility to come into contact).
- b = starting sphere
radius (in Å) - simulations are started by
placing the center of the
second solute at distance b
from the first solute's center. For
calculation of association constants, there should be
no interactions between the solutes at this distance. With
finite size grids, b can be
selected to be larger than the sum of the solute 1 (or 2) grid size
and solute
2 (or 1) size .
- c = end sphere radius - simulation of trajectories stop at separation c between solutes' centers. >
- rboost = boost value to
move the solutes apart when more than novers
moves were not accepted due to overlaps. Boosting is introduced
to avoid infinite trajectories and can influence
simulation results, because trajectories are modified by boosts.
- novers = number of overlaps allowed before making a boost
- djump
= threshold for the move length - moves in one timestep greater than
this value are counted (switched off)
- icommrxn = reaction condition switch
- dind = independence distance for icommrxn=3 (in Å)
# BLOCK 09: win0, dwin, nwin, iwrec, nwrec, ncec, rxna1f, rxna2f
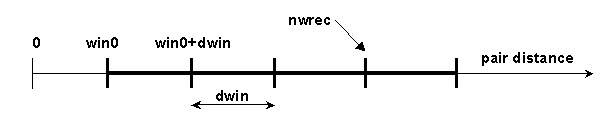
- win0 = reaction distance
window minimal value (in Å)
- dwin = reaction distance window step (in Å).
- nwin = number of reaction distance windows.
- iwrec = switch (0 or 1) to decide if complexes satisfying reaction conditions should be recorded
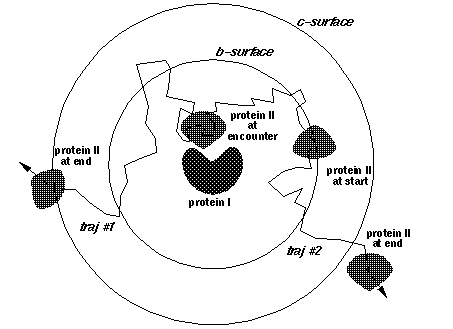
- nwrec = number of the window at which the complexes are recorded. For example, in the situation below, nwin=5 reaction distance windows are defined, and rates for forming contacts at each of these 5 distances will be monitored. Encounter complexes with contact distances less than win0+dwin*(nwrec-1), will be recorded if iwrec is set to 1. E.g. in the figure below nwrec=4 is represented. This parameter is only relevant to writing complexes, when iwrec=1.
- ncec = number of contacts
to define the encounter complex, set to 2 if not 1,2 or 3. This
parameter is only relevant for writing complexes when iwrec=1.
- rxna1f = filename for
solute 1 reaction atoms - reaction atom
pairs are
formed by atoms of solutes 1 and 2 from the lines with the same number,
i.e. atom on the line N in the file rxna1f forms a pair with the atom
on the line N in the file rxna2f. Reaction atoms do not have to
be
real atoms, although in this file these atoms/points should be given
like atoms in
PDB format. Different combinations of the same
atoms/points can be prepared by editing these reaction atom files.
- rxna2f = filename for solute 2 reaction atoms.
# BLOCK 10: epfct, ep1f, ep2f, qef1f, qef2f
- epfct = factor by which the
electrostatic potential grid
values read-in are to be multiplied. Should be 1 if no modifications are needed.
- ep1f = filename for the electrostatic potential of solute 1 in UHBD binary format. It is assumed that the potentials are written in kcal/mol/e units. Note that APBS generated grids written in UHBD format are in units of kT/e and ascii and as such cannot be used directly with SDA. They should be rescaled and converted with the auxiliary program apbs2uhbd, to bring them into consistency with UHBD. >
- ep2f = filename for the
electrostatic potential of solute 2 in
UHBD
format, binary.
- qef1f = filename for
effective charges of solute 1. This is output of the ECM
program. The
effective charge presentation in variant R (extension _R) is to be
used, see detailed description here.
- qef2f = filename for effective charges of solute 2
- edfct = factor by which the
electrostatic desolvation potential
grid values are to be multiplied by. The value 1.00 corresponds to the uncorrected
electrostatic desolvation penalty for a charge in a solvent due to the low
dielectric cavity of a solute treated as a collection of van der Waals
spheres. More appropriate factors can be derived by comparing SDA
calculated energies of recorded complexes with energies calculated with
more accurate methods (solving the finite difference Poisson-Boltzmann
equation, for example).
- ed1f = filename for the
electrostatic desolvation potential of
solute 1
in UHBD format. This grid can be calculated with program mk_ed_grd
which is included in SDA distribution.
- ed2f = filename for the electrostatic desolvation potential of solute 2 in UHBD format
- hdfct = factor by which the hydrophobic desolvation potential grid values are to be multiplied by. In the SDA program, hydrophobic desolvation energies are calculated by multiplying the solvent accessibilities of the atoms of one of the solutes by the value of the hydrophobic desolvation potential grid calculated for the other solute. If the grid is calculated so that the sum of contributions from all accessible atoms gives the buried solvent accessible area, then realistic values for this factor are in the range 0.005-0.060 kcal/mole/Å2 (or 5-60 cal/mole/Å2). The value of this factor can also be derived by comparing SDA calculated energies of recorded complexes with energies calculated with more accurate methods.
- hd1f = filename for the hydrophobic desolvation potential of solute 1 in UHBD format. This grid can be calculated with program mk_hd_grd which is included in SDA distribution. >
- hd2f = filename for the hydrophobic desolvation potential of solute 2 in UHBD format
- fnintr = name of the file where intermediate results (fraction of reactive trajectories for every reaction window distance and for forming 1, 2, 3 or 4 contacts at these distances) are written every nprint run.