simulation of diffusionalassociation of proteins
(version 4.23)
EMBL, Heidelberg
February 2000
The description of each input variable follows:
dseed - random number generator seed. Defines the sequence of random numbers used to model the Brownian dynamics. Different numbers result in different sampling and trajectories.
nrun, nprint, iwrite - the number of Brownian dynamics runs, frequency of printout of reaction events, and the number of the trajectory to write.
probes, thres, hexcl, dsfactor - the probe radius used to represent the atoms of the second protein and the threshold value for atomic accessibility. The atoms of protein 2 having accessibilities less than thres are not used for determinimg molecular exclusion in the simulations. hexcl is the spacing for the exclusion grid of the protein 1. dsfactor is a factor to multiply the desolvation penalty grid (see the description of the desolvation term).
pdb1f, pdb2f - the names of files with atomic coordinates of proteins 1 and 2, in pdb format ('ATOM',a24,3f8.3).
po1f, po2f - the names of files with electrostatic potentials of proteins 1 and 2. UHBD binary format.
qef1f, qef2f - the names of files with charges for proteins 1 and 2. ('ATOM',24x,3f8.3,f15.8).
rxna1f, rxna2f - the names of files with reaction atom coordinates for proteins 1 and 2 in pdb format. ('ATOM',24x,3f8.3).
ic1fix, xc1 - 2 lines, the integer on the first line is 1 or 0, the second line defines the coordinates of the center of the first protein supplied by the user. These coordinates are used by the program when ic1fix=1, and recomputed when ic1fix=0.
ic2fix, xc2 - the same as above for the second protein center.
icomm - switches the encounter complex modeling options. icomm=1-3 deal with 3 different ways to count reaction events: icomm=1 counts RMSD reaction conditions (Fig 1),
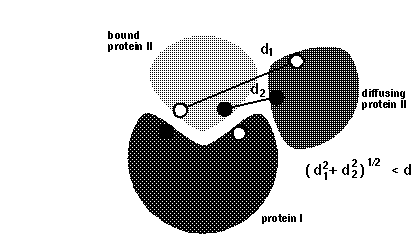
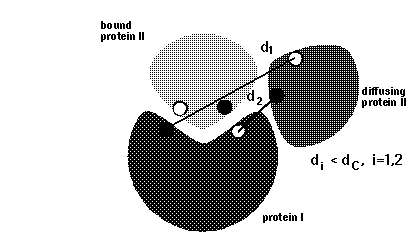
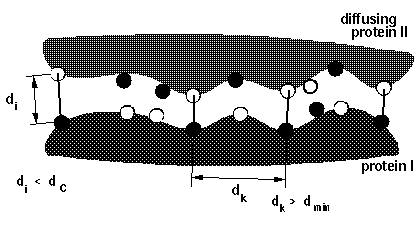
Note that more reaction criterion atoms and more sophisticated reaction criteria imply longer computation times, e.g. sda computations may take 3 times longer when option icomm=3 with 88 reaction criterion atoms is chosen compared to using option icomm=2 with 7 reaction criterion atoms.
iforce=0 switches off all the interaction forces except the exclusion forces. iforce =1 is the standard option when the forces are computed using the potential grid of protein 1 (2) and the charges of protein 2 (1). iforce =2 does not use the potential grids at all but computes the forces using the charges alone - this is efficient only when the number of charges is less than 10. Charge-charge treatment is useful when a large executable is not affordable as a separately compiled small executable can be used. For iforce =2, it is necessary to give a correct value of aiostr == ionic strength of the solvent in mM.
dm, dr, drI , irot2f - 3 diffusion coefficients: relative translational, rotational for protein 2, rotational for protein 1. irot2f=0 switches off the modeling of rotation of the first protein. dm is in Å2/ps, dr and drI are in radian2/ps.
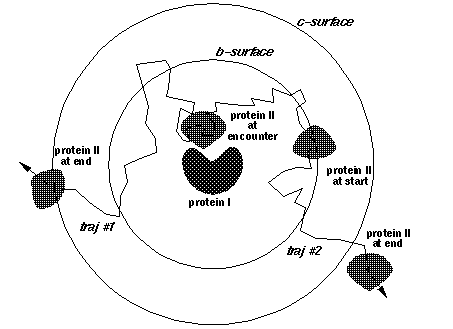
b, c - the distances at which the trajectories are started and truncated, respectively (Fig. 4)
rboost, novers - after novers rejections of Brownian dynamics moves, the proteins are taken apart from one another by a distance rboost. The number of boosts is reported in the output log. Choice novers ~100 usually does not need any boosts, unless the interactions are too strong due to use of an unrealistically small probe size. Many boosts may indicate erroneous input (uncorrelated pdb and grid etc).
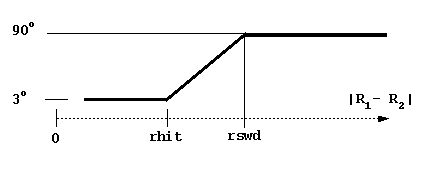
dt1, swd1, dt2, swd2, rswd - time step parameters. dt1 is the smallest time step to be used, it is automatically used when the proteins are close to overlap. The next 3 parameters define the time step profile as in Figure 4:
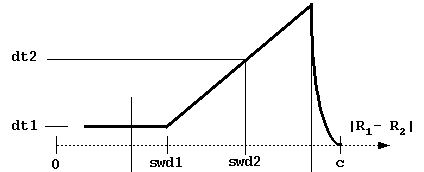
The decreasing of time step to 0 near the c-surface is done automatically to reduce artifacts due to trajectory truncations. Although the profile of the time step influences the computed rates, the most important parameter is dt1. Take care not to choose this parameter too small (since the computing time scales almost linearly by it). The physical parameter sqrt(dt*dm) measures an average Brownian dynamics step length. This must be less than 1 Å, which is a characteristic distance of every protein surface roughness since the atomic bond lengths and van der Waals radii define this scale. Moreover, it should be less than the characteristic distance over which the interaction forces vary substantially, which means that the optimal value is influenced by interaction force strength: one needs smaller time steps for stronger interacting proteins. The rotations are accumulated when the time step is small to make the simulations faster. The rotation is carried out only when the accumulated random component of rotation is ~ 3o at small interprotein separations and ~90o at separations larger than rswd (see figure).
win0, dwin, nwin - the reaction complex will be recorded for each of nwin contacts (or RMSD distances) di, i=1,nwin. di=win0+(i-1)*dwin. If iwrec.ne.0 then the positions of the second protein will be written to file fort.55, when 2 contacts at distance dnwrec are satisfied.
dspo1f, dspo2f - the names of files with charge desolvation penalties of proteins 1 and 2. UHBD binary format. Charge desolvation free energies are computed by a separate program in sda distribution, mk_ds_grd.f. The charge desolvation penalty for protein 1 is the sum of desolvation penalties of each charge of the protein 1, i=1,...; the desolvation penalty of each charge is the sum of desolvation penalties due to the low dielectric cavity of each atom of protein 2, i.e. the final expression giving the desolvation penalty for protein 2 due to the presence of protein 1 is:
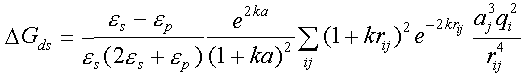
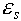 and
and 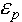 are the dielectric constants of the solvent and the protein, respectively;
k
- is the Debye-Hueckel parameter, qi is the value of
the i-th charge on the protein 2, aj - radius of the
j-th atom of the protein 1, a is the sum of the average radius of
the atoms of the protein 1 incremented by the ionic radius of salt (~1.7+1.5=3.2
Å). This description is approximate and depends on the treatment
of the interior dielectric of the protein and the method to define the
dielectric boundary between the protein and solvent. Meaningful values
are 1.62 when the protein interior dielectric is 4 and the dielectric boundary
between the protein interior and high dielectric solvent is defined as
van der Waals surface of the protein. The value of ca 4.0 would approximate
the desolvation penalties when the molecular surface of the protein is
used as a dielectric boundary, although in this case the description of
charge desolvation via precalculated grids is much less accurate.
are the dielectric constants of the solvent and the protein, respectively;
k
- is the Debye-Hueckel parameter, qi is the value of
the i-th charge on the protein 2, aj - radius of the
j-th atom of the protein 1, a is the sum of the average radius of
the atoms of the protein 1 incremented by the ionic radius of salt (~1.7+1.5=3.2
Å). This description is approximate and depends on the treatment
of the interior dielectric of the protein and the method to define the
dielectric boundary between the protein and solvent. Meaningful values
are 1.62 when the protein interior dielectric is 4 and the dielectric boundary
between the protein interior and high dielectric solvent is defined as
van der Waals surface of the protein. The value of ca 4.0 would approximate
the desolvation penalties when the molecular surface of the protein is
used as a dielectric boundary, although in this case the description of
charge desolvation via precalculated grids is much less accurate.
The file fort.66 contains the length of each trajectory.
The file fort.77 contains the values of electrostatic potentials of protein 1 and protein 2 along 5 selected lines in 3-dimensional space; the values of the desolvation penalty grid along the same 5 lines; a section through the exclusion grid in ascii format in the plane passing through the center of the protein 1; reaction atom information.
The file fort.99 lists occurrence of reaction atom contacts. The first record is for the number of contacts observed at each specified contact distance for each reaction atom pair. The second record gives the number of contacts counted only once per Brownian dynamics run.
Since the above files are overwritten each program run, one needs to rename them after the execution if they are needed.
The file runs.done has the number of runs performed so far. The information is written every 50 runs and is useful when the printout frequency is low and the machine writes to files with buffering.
Standart output (fort.6) has the following.
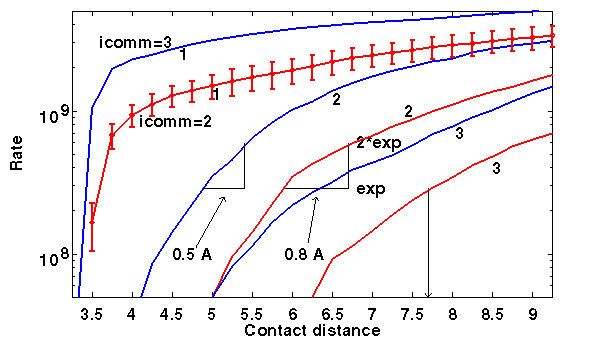
It is necessary to assume too small 1-contact distance in order to predict experimental association rates. In this case, they are 3.5-3.75 Å, i.e. close to the minimal atom-atom distance allowed in the model. This means that a 1-contact description of the encounter complex is impossible. Moreover, the results are sensitive to definition of contact distance. 2-3 contacts in either interresidue or independent contacts model are meaningful. 2 interresidue contacts happen at 6.0-6.7 Å and 2 independent atom-atom contacts - at 4.0-4.5 Å. The left and right interval ends define the contact distances giving the rates equal to experimental (exp) and twice experimental (2*exp) rates, respectively.
February, 2000