averaging Boltzmann factor within specified reaction distance
version 4.23
EMBL, Heidelberg
February 2000
The description of each number follows:
dseed - random number generator seed. Defines the sequence of random numbers used to model the Brownian dynamics. Different numbers result in different sampling and trajectories.
nrun - the number of random angles at every point of the sampling grid to be tried to compute the average Boltzmann factor.
probes, thres, hexcl, dsfactor - the probe radius used to represent the second protein atoms and the threshold value for atomic accessibility. The atoms of protein 2 having accessibilities less than thres are not used in computing exclusion. hexcl is the spacing for the exclusion grid of protein 1. dsfactor is a factor to multiply the desolvation penalty grid (see the description of the desolvation term).
pdb1f, pdb2f - the names of files with atomic coordinates of proteins 1 and 2, in pdb format ('ATOM',a24,3f8.3). The first protein file may have a record giving the scaling factor for its radius, after the coordinate record: ('ATOM',24x,3f8.3,f15.8) when the sdabfsc program is used. If no record is present, then the factor is 1.0.
po1f - the name of the file with the electrostatic potential of protein 1. UHBD binary format. po2f - not used here, but it is read in.
qef2f - the names of files with charges for proteins 1 and 2. ('ATOM',24x,3f8.3,f15.8). qef1f - not used here, but it is read in.
rxna1f, rxna2f - the names of files with reaction atom coordinates for proteins 1 and 2 in pdb format. ('ATOM',24x,3f8.3).
ic1fix, xc1 - 2 lines, the integer on the first line is 1 or 0, the second line defines the coordinates of the center of the first protein supplied by the user. These coordinates used by the program when ic1fix=1, and recomputed when ic1fix=0.
ic2fix, xc2 - the same as above for the second protein center.
icomm - switches the encounter complex modeling options. icomm=1-3 deal with 3 different ways to count reaction events: icomm=1 counts RMSD reaction conditions icomm=2 interresidue contact pairs, icomm=3counts independent contact pairs among all possible donor-acceptor pairs formed between the 2 proteins. Refer to readme for Brownian dymnamics program.
iforce =0 switches off all the interaction forces except the exclusion forces. iforce =1 is the standard option when the forces are computed using the potential grid of protein 1 (2) and the charges of protein 2 (1). iforce =2 does not use the potential grids at all but computes the forces using the charges alone - this is efficient only when the number of charges is less than 10. Charge-charge treatment is useful when a large executable is not affordable as a separately compiled small executable can be used. For iforce =2, it is necessary to give the correct value of aiostr == ionic strength of the solvent in mM.
dm, dr, drI , irot2f - not used here.
dt1, swd1, dt2, swd2, rswd - not used here.
win0, dwin, nwin - the reaction
complex will be recorded for each of nwin contacts (or
RMSD distances) di, i=1,nwin. di=win0+(i-1)*dwin.
If iwrec.ne.0 then the positions of the second protein will be written
to file fort.55, when 2 contacts at distance dnwrec
are satisfied.
dspo1f, dspo2f - the names of
files with charge desolvation penalties of proteins 1 and 2. UHBD binary
format. dspo2f - is not used, but read in.Charge
desolvation free energies computed by a separate program in sda distribution
mk_ds_grd.f.
See description of charge desolvation in sda.html.
igwrite - Option to write grids
or not: igwrite=1 - the program will calculate and write
grids, iform=0 - only the table of Boltzmann factors as
a function of contact distance is written.
hs,nsx,nsy,nsz - the parameters
of the sampling grid: the program will make nrun samplings of random angles
at every point of the grid, compute reaction conditions and add up the
Boltzmann factors if the conditions are satisfied, thus creating the table
of Boltzmann factors at every reaction distance. hs -
spacing of the sampling grid, nsx,nsy,nsz
- its dimensions.
It is centered at the center of the second protein, given in pdf2f.
It has to be large enough that all important interaction regions are included
(to get the complete sampling for the Boltzmann factor grids as well as
to have correct Boltzmann factors at each reaction distance).
psof,psofe,iform - The names
of the grids to write the orientational Boltzmann factor average and Boltzmann
averaged electrostatic interaction energy and their format (iform=1
- ascii, iform=0 - binary UHBD format).
The file fort.66 contains the length of each trajectory.
The file fort.77 contains the electrostatic potentials of protein 1 and protein 2 along 5 selected lines in 3-dimensional space; a section through the exclusion grid in ascii format in the plane passing through the center of protein 1; reaction atom information.
Since the above files are overwritten each program run, one needs to rename them after the execution if they are needed.
The file runs.done has the number of runs (orientational samplings) performed so far. The information is written every 50 runs and is useful when the printout frequency is low and the machine writes to files with buffering.
Standard output (fort.6) has the following.
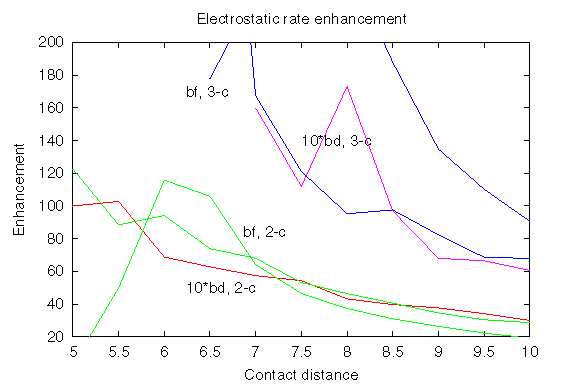
Here the rate enhancements are shown, which were obtained by computing Boltzmann factors (marked bf) for the encounter complexes defined by 2 or 3 contacts at the distance specified on the x-axis; there are 2 curves for each Boltzmann factor, which correspond to 2 different ways of sampling the Boltzmann factors. The rate enhancements obtained by BD simulations are marked bd, and by the number of contacts, 2 or 3. In fact, the BD rate enhancements are multiplied by 10 to bring the 2 estimates into quantitative correspondence. Consequently, the ratio of rate enhancements for encounter complexes defined by 2 and 3 contacts is reproduced reasonably well by sampling of the Boltzmann factor. The Boltzmann factor overestimates by a factor of 10 the rate enhancement obtained by BD. Partially, this overestimation is due to applying a cutoff to the interactions at a 54 Å distance of (Protein I centre)-(Protein II charge). The sampling of the Boltzmann factor is insensitive to this cutoff. On the other hand, BD simulations are sensitive to this cutoff, although, this cutoff contribution to rate enhancement has been shown not exceed a factor of 2.
Viel Spass.
February 2000