ACETONE

| electrostatic energies between acetone and solvent during MD equilibration [kJ/mol] | |||
|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP
| |
| CHARMM | -49.8 | -29.4 | -15.6 |
| OPLS_SPCE | -64.6 | -39.1 | -16.4 |
| OPLS_TIP3P | -58.4 | -34.6 | -18.4 |
| CVFF | -65.0 | -38.7 | -19.1 |
| GROMOS87 | -62.0 | -34.2 | -16.3 |
| GROMOS_690 | -56.4 | -34.0 | -14.8 |
| GROMOS_POL | -55.2 | -31.2 | -15.1 |
| Lennard Jones energies between acetone and solvent during MD equilibration [kJ/mol] | |||
|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP
| |
| CHARMM | -48.1 | -50.2 | -51.6 |
| OPLS_SPCE | -29.1 | -33.4 | -35.6 |
| OPLS_TIP3P | -28.3 | -32.1 | -33.9 |
| CVFF | -37.4 | -41.1 | -43.3 |
| GROMOS87 | -40.0 | -44.1 | -46.9 |
| GROMOS_690 | -28.6 | -31.4 | -34.4 |
| GROMOS_POL | -25.2 | -28.9 | -31.7 |
| deltaGhydr (longest simulation for each system) [kJ/mol] | |||
|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP
| |
| CHARMM | -28.4 | -18.9 | -12.3 |
| OPLS_SPCE | -19.2 | -5.5 | +2.8 |
| OPLS_TIP3P | -16.5 | -8.1 | -2.7 |
| CVFF | -28.2 | -17.1 | -8.3 |
| GROMOS87 | -34.0 | -24.5 | -15.1 |
| GROMOS_690 | -17.9 | -6.3 | +1.0 |
| GROMOS_POL | -13.2 | -1.7 | +4.4 |
DIMETHYL ETHER

| electrostatic energies between dimethyl ether and solvent during MD equilibration [kJ/mol] | |||
|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP
| |
| CHARMM | -23.3 | -14.2 | -7.8 |
| OPLS_SPCE | -31.0 | -17.9 | -7.9 |
| OPLS_TIP3P | -28.3 | -17.0 | -8.2 |
| CVFF | -31.3 | -17.7 | -9.0 |
| GROMOS87 | -24.4 | -12.7 | -6.3 |
| GROMOS_690 | -22.7 | -13.7 | -6.0 |
| GROMOS_POL | -23.0 | -12.8 | -5.5 |
| Lennard Jones energies between dimethyl ether and solvent during MD equilibration [kJ/mol] | |||
|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP
| |
| CHARMM | -41.6 | -41.2 | -42.3 |
| OPLS_SPCE | -24.0 | -26.9 | -28.3 |
| OPLS_TIP3P | -23.9 | -26.4 | -27.3 |
| CVFF | -33.3 | -35.7 | -36.0 |
| GROMOS87 | -37.7 | -39.1 | -40.2 |
| GROMOS_690 | -26.9 | -27.1 | -29.5 |
| GROMOS_POL | -24.0 | -25.6 | -26.9 |
| deltaGhydr (longest simulation for each system) [kJ/mol] | |||
|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP
| |
| CHARMM | -12.2 | -7.0 | -5.6 |
| OPLS_SPCE | +1.5 | +3.4 | +5.7 |
| OPLS_TIP3P | -2.0 | +2.3 | +5.2 |
| CVFF | -11.1 | -4.6 | -2.9 |
| GROMOS87 | -17.4 | -13.3 | -11.7 |
| GROMOS_690 | -3.2 | +2.2 | +3.5 |
| GROMOS_POL | +1.8 | +5.4 | +8.5 |
PROPANE
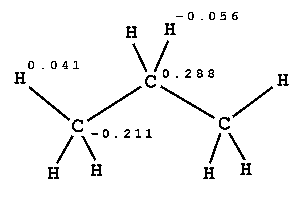
| electrostatic energies between propane and solvent during MD equilibration [kJ/mol] | ||||
|---|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP | 0 charges
| |
| CHARMM | -0.3 | -0.2 | -0.2 | |
| OPLS_SPCE | -0.3 | -0.2 | -0.1 | |
| OPLS_TIP3P | -0.5 | |||
| CVFF | -0.3 | -0.2 | -0.1 | |
| GROMOS87 | 0 | |||
| GROMOS_690 | 0 | |||
| GROMOS_POL | 0 | |||
| Lennard Jones energies between propane and solvent during MD equilibration [kJ/mol] | ||||
|---|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP | 0 charges
| |
| CHARMM | -46.5 | -46.6 | -46.4 | |
| OPLS_SPCE | -31.4 | -30.9 | -29.8 | |
| OPLS_TIP3P | -30.2 | |||
| CVFF | -41.8 | -42.2 | -41.5 | |
| GROMOS87 | -47.7 | |||
| GROMOS_690 | -32.9 | |||
| GROMOS_POL | -29.8 | |||
| deltaGhydr (longest simulation for each system) [kJ/mol] | ||||
|---|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP | 0 charges
| |
| CHARMM | -1.6 | -0.9 | -1.4 | |
| OPLS_SPCE | +12.1 | +13.2 | +9.1 | |
| OPLS_TIP3P | 10.2 | |||
| CVFF | +1.5 | +2.2 | -0.1 | |
| GROMOS87 | -11.7 | |||
| GROMOS_690 | +7.6 | |||
| GROMOS_POL | +15.1 | |||
CYCLO-PROPANE
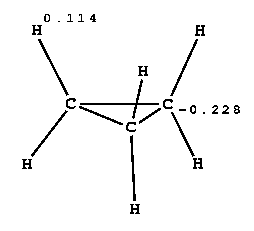
| electrostatic energies between cyclopropane and solvent during MD equilibration [kJ/mol] | ||||
|---|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP | 0 charges
| |
| CHARMM | -3.2 | |||
| OPLS_SPCE | -3.1 | |||
| OPLS_TIP3P | -3.2 | |||
| CVFF | -2.2 | |||
| GROMOS87 | 0 | |||
| GROMOS_690 | 0 | |||
| GROMOS_POL | 0 | |||
| Lennard Jones energies between cyclopropane and solvent during MD equilibration [kJ/mol] | ||||
|---|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP | 0 charges
| |
| CHARMM | -40.6 | |||
| OPLS_SPCE | -27.5 | |||
| OPLS_TIP3P | -25.9 | |||
| CVFF | -38.2 | |||
| GROMOS87 | -45.6 | |||
| GROMOS_690 | -31.8 | |||
| GROMOS_POL | -28.8 | |||
| deltaGhydr (longest simulation for each system) [kJ/mol] | ||||
|---|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP | 0 charges
| |
| CHARMM | -3.0 | |||
| OPLS_SPCE | +11.1 | |||
| OPLS_TIP3P | +9.7 | |||
| CVFF | -0.5 | |||
| GROMOS87 | -11.9 | |||
| GROMOS_690 | +6.7 | |||
| GROMOS_POL | +15.6 | |||
CAMPHOR
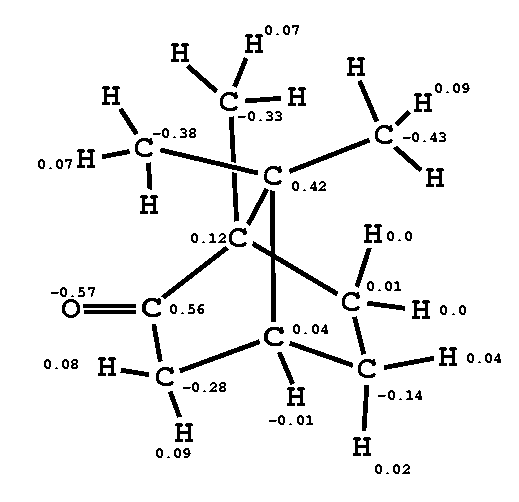
| electrostatic energies between camphor and solvent during MD equilibration [kJ/mol] | |||
|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP
| |
| CHARMM | -46.5 | -28.4 | -15.0 |
| OPLS_SPCE | -63.5 | -37.9 | -17.7 |
| OPLS_TIP3P | -58.3 | -33.0 | -16.7 |
| CVFF | -62.0 | -34.2 | -19.1 |
| GROMOS87 | -65.6 | -36.6 | -18.5 |
| GROMOS_690 | -62.6 | -36.2 | -18.5 |
| GROMOS_POL | -60.5 | -35.7 | -17.4 |
| Lennard Jones energies between camphor and solvent during MD equilibration [kJ/mol] | |||
|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP
| |
| CHARMM | -99.0 | -103.8 | -106.0 |
| OPLS_SPCE | -68.9 | -71.6 | -75.1 |
| OPLS_TIP3P | -65.1 | -68.2 | -71.5 |
| CVFF | -90.0 | -94.7 | -95.5 |
| GROMOS87 | -105.5 | -108.4 | -111.3 |
| GROMOS_690 | -74.9 | -78.0 | -81.9 |
| GROMOS_POL | -67.4 | -71.7 | -75.0 |
| deltaGhydr (longest simulation for each system) [kJ/mol] | |||
|---|---|---|---|
| 1.0 x ESP | 0.8 x ESP | 0.6 x ESP
| |
| CHARMM | -41.6 | -27.5 | -26.8 |
| OPLS_SPCE | -23.9 | -3.7 | +1.6 |
| OPLS_TIP3P | -17.7 | -14.2 | -3.2 |
| CVFF | -44.0 | -33.6 | -28.0 |
| GROMOS87 | -66.2 | -52.2 | -40.2 |
| GROMOS_690 | -29.2 | -17.3 | -11.3 |
| GROMOS_POL | -22.6 | -10.9 | -3.3 |
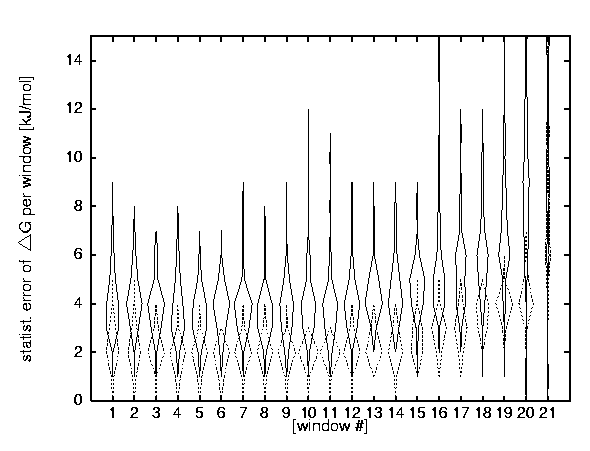
STATISTICAL ERRORS
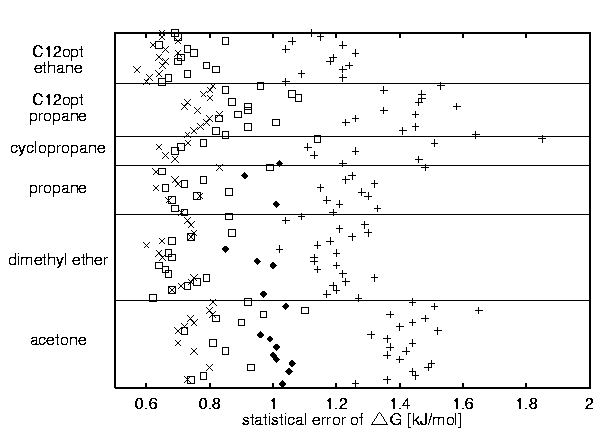
A + B stands for windows of A equilibration steps and B data acquisition steps, A + (B < x < C) stands for windows of A equilibration steps and between B and C data acquisition steps :
(+) 1000 + 5000, (filled diamond) 1000 + (5000 < x < 30000) , (open square) 6000 + 15000, and (cross) 6000 + (15000 < x < 30000)
AVERAGE STATISTICAL ERROR PER WINDOW
Volkhard Helms
Rebecca Wade
15 November 1996