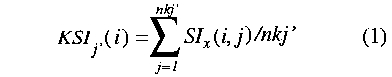
where SIx are Carbo (c) [41] or Hodgkin (h) similarity indices [42] and nkj'= number of compounds in the kernel of class j'.
Keywords:QSAR, auxins, molecular modeling, molecular alignment,
interaction similarity, n-alkyl-IAA, CHIME, MAGE
Abbreviations: MIF: molecular interaction field; IAA: indole-3-acetic acid; SI: similarity index; Me: methyl; Et: ethyl; Pr: propyl; Bu: butyl
2.Outline of the classification method
3. Classification of new compounds
5. Distribution of compounds
in 3D space defined by similarity of their interaction properties
6. Summary
(1) Model molecular structures
(2) Align conformers either by using atomic properties
(program SEAL [39])
or by using interaction properties on the probe
accessible surface of the molecule
(3) Calculate molecular interaction fields (MIF) using the GRID program [40]
(4) Divide compounds into classes according to similarity
indices [41, 42] computed for their
MIFs.
For auxin-related compounds, the classes are defined
as follows. Class 1: active compounds-auxins, class 2: very low activity
compounds with antiauxin characteristics, class 3: inactive compounds,
class 4: antiauxins, i.e. compounds with inhibitory characteristics.
(5) Classify "new" compounds according to the similarity
between their MIFs and the MIFs of "class kernels". A
"kernel" is a reduced set of compounds defining the class.
Although classes were on average distinguishable in both sets of conformations, better differentiation was achieved for the "T" than the "P" conformers [1]. So for the classification of any new compound, low energy conformers similar to the tilted conformation of IAA are used. The "T" conformers of all analyzed compounds are shown in the 3D library of auxins. The conformers may be obtained using any available ab initio, semiempirical or molecular mechanics method that enables adequate modeling of the desired compound. Although none of the molecular mechanics force fields that we tested was able to accurately reproduce results obtained by ab initio calculations (RHF 6-31G*) [34, 35, 36], our experience [36] shows that the CFF91 force field [37] used in the DISCOVER program [38] gives sufficiently good results.
Calculation of MIFs:
Molecular interaction fields were
computed for each compound using the GRID [40] program.
Calculations were performed either on points on a 1
spacing grid in a volume around molecules or on points defining
a probe accessible surface. The
most suitable probes were found to be H2O, NH2+,
CH3 and carbonyl O [1].
Biological activity
According to the calculated KSIs, both
conformers of 4-, 5- and 6-Et-IAA, both methyl substituted IAAs, 6- and
7- Me-IAA as well as 5-Pr-IAA and 5-Bu-IAA belong to class 1. The same
classification was determined before for the "T" and "P" conformers of
4-Et-IAA and 4- and 5-Me-IAA [1]. The results obtained are in agreement
with the available experimental data [2, 3,
6, 13, 19, 20,
28, 29]. In Avena coleoptile
straight growth tests [2, 3, 19,
29, 42], the ring substituted Me-IAAs,
4-Et-IAA, 5- Et-IAA, 5-n-Pr-IAA and 5-n-Bu-IAA enhance growth.
In addition the binding measurements [13] show that
4-Et-IAA and 2-Me-IAA bind to auxin binding protein 1, a putative auxin
receptor [48-50], although the binding affinity of 2-Me-IAA
is very low. 2-Et-IAA is predicted to be an auxin,
as it was mostly classified in class 1. However, its KSI1 is
lower than those of the majority of the other compounds and in three cases
it was classified differently: once in class 4, and twice in class 3. Koegl
and Kostermans [44] claimed 2-Et-IAA to be inactive.
However, this experiment was done in 1935 and the compound might not have
been correctly identified when synthesized as a later attempt to synthesize
this compound by the procedure given failed [31].
A new experiment for this compound is planned for the near
future [31].
By classification procedures, 7-Et-IAA is predicted to
have weak antiauxin characteristics. The biological
activity of this compound has not been measured yet.
The other IAA derivatives
5-Methoxy-IAA was mostly classified
into class 1, except once, in class 3 and once, in class 2. Findlay
and Dougherty [43] determined moderate auxin activity
for it.
4,5-Cl2-IAA is predicted
to be an auxin in agreement with the recently performed Avena coleoptile
straight growth tests on di-chloro substituted IAAs [30],
by which 4,5-Cl2-IAA is one of the most active auxins.
For 5-Br-IAA, Bttgeret al. [22]
measured moderate auxin activity in Avena coleoptile, and by our
method it is classified in class 1.
The
mean KSIi (i=1- 4) values, for the H2O (#1) and
NH2+ (#2) probes, calculated from three sets of differently
aligned MIFs on the grid are represented as histograms
with their standard deviations given by bars.
The compounds 1-12 are ordered as follows: 4-Et-IAA (1),
5-Et-IAA (2), 6-Et-IAA (3), 7-Et-IAA (4), 7-Me-IAA (5), 6-Me-IAA (6), 5-Br-IAA
(7), 5-n-Bu-IAA (8), 4,5-Cl2-IAA (9), 5-methoxy-IAA (10), 5-n-Pr-IAA
(11) and 2-Et-IAA (12). The histograms for 7-Et-IAA and 2-Et-IAA
are colored differently.
The results on the probe accessible surface obtained with MIFs for the CH3 probe.
additional
set (including compounds analysed
before [1] and in this work)
Compounds from this set were classified using the proposed
The analysis of the distribution of
similarity indices in 3D space by MAGE is a new approach to the classification
problem. It enables better rationalization of correlations between
the classes.
The distance between compounds
is defined as D(i,j) = sqrt(1-SI(i,j)).
Four points defining the 3D space
were chosen as follows:
a) The first two points (1 and
2) are chosen as those for which the distance D(1,2), is
largest. This D(1,2) is then used for
normalization of all other distances.
b) Point 3 is chosen so that the
area of the triangle formed by points 1,2 and 3 is the largest.
c) Point 4 is chosen so that the
volume of the tetrahedron formed by points 1,2,3 and 4 is the largest.
The 3D coordinates assigned
to these 4 points are (0,0,0), (1,0,0), (a,b,0), (c,d,e) , where a, b,
c, d and e are computed
unambiguously from the 6 pairwise distances. The
other compounds are projected onto this defined 3D space with coordinates
determined from
their SI(i,j) values.
These projections
are visualized using the MAGE program. Distributions are based on SI(i,j)
of MIFs determined for the probes H2O
(#1),
NH2+ (#2),
CH3 (#3)
and
carbonyl
O (#4).
In these distributions,
three main regions can be distinguished, those populated with auxins, those
with inhibitors and those with inactive compounds. The region occupied
with auxin-related compounds without growth promoting activity is
clearly separated from the others for all probes. However, although most
of the antiauxin compounds are far from auxins, the border between these
two populations is not always very clear. The compounds from class 2 are
situated around the border between classes 1 and 4.
2. Porter, W.L. and Thimann, K.V., Phytochemistry, 4 (1965) 229.
3. Kojic-Prodic, B., Nigovic, B., Tomic, S., Ilic,
N., Magnus, V., Konjevic, R., Giba, Z. and
Duax, W.L., Acta Crystallogr., B47 (1991) 1010.
4. Nigovic, B., Kojic-Prodic, B., Antolic, S., Tomic,
S., Puntarec, V. and Cohen, J.D.,
Acta Crystallogr., B52 (1996) 332.
5. Antolic, S., Kojic-Prodic, B., Tomic, S., Nigovic,
B., Magnus, V. and Cohen, J.D.,
Acta Crystallogr., B52 (1996) 651.
6. Lutz, B. T. G., van der Windt, E., Kanters, J.,
Klmbt, D., Kojic-Prodic B. and Ramek, M.,
J. Mol. Struct., 382 (1996) 177.
7. Ray P. M., Dohrmann U. and Hartel R., Plant Physiol., 60 (1977) 585.
8. Hatano T., Katayama M. and Marumo S., Experientia, 43 (1987) 1237.
9. Hatano T., Kato Y., Katayama M. and Marumo S., Experientia, 45 (1989) 400.
10. Katayama M., Kato Y., Kimoto H., Fuji S., Experientia, 51 (1995) 721.
11. Katekar G. F. and Geissler A. E., Phytochemistry, 21 (1982) 257.
12. Katekar G. F. and Geissler A. E., Phytochemistry, 22 (1983) 27.
13. Rescher U., Walther A. Schiebl C. and Klmbt D., J. Plant Growth Regul., 15 (1996) 1.
14. Reinecke D. M., Ozga J. A. and Magnus V., Phytochemistry, 40 (1995) 1361.
15. Wain R. L. and Wightman F., Ann. Appl. Biol.,40 (1953) 244.
16. Fawcett C. H., Wain R. L. and Wightman F., Ann. Appl. Biol., 43 (1955) 342.
17. Veldstra H. and Van der Westeringh C., Recl. Trav. Chim. Pays. Bas., 70 (1951) 1113.
18. Brustrm H., Physiol. Plant., 3 (1950) 277.
19. Nitsch, J. P. and Nitsch, C., Plant Physiol., 3 (1956) 94.
20. Muir R. M. and Hansch C., Physiol. Plant., 28 (1953) 218.
21. Stenlid G. and Engvild K. C., Physiol. Plantarum, 70 (1987) 109.
22. Bttger, M. Engvild, K. C. and Soll, H., Planta ,140 (1978) 89.
23. Toothill J. R., Wain R. L. and Wightman F., Ann. Appl. Biol., 44 (1956) 547.
24. Hansen B., I. Bot. Nat. (Lond.), (1954) 230.
25. Veldstra H., Rec. Trav. Chim. Pays-Bas, 71 (1952) 15.
26. Pybus M. F., Wain R. L. and Wightman F., Nature, 182 (1958) 1094.
27. Smith G., Kennard, C. H. L. and White, A. H., Acta Crystallogr.,B34 (1978) 2885.
28. Hoffmann, A. L., Fox, S. W. and Bullock, M.W., J. Biol. Chem., 196 (1952) 437.
29. Antolic S., Kojic-Prodic B., Magnus V.,
Salopek B., Tomic S.:
Plant Physiology 114 (3,
Suppl.) 158 (1997).
30. Antolic S., Salopek B., Kojic-Prodic B., Magnus
V., and Cohen J. D., Structural
characterization and
auxin properties of dichlorinated indole-3-acetic acids.
Plant Growth
Regulation (1998), accepted for publication.
31. Magnus V., private communication
32. Chemscape CHIME, http://www.mdli.com/chemscape/chime
33. MAGE_3.1, http://www.faseb.org/protein/kinemages/MageSoftware.html
34. Ramek, M., Tomic, S. and Kojic-Prodic, B., Int.
J. Quant. Chem.: Quant. Biol. Symp.,
22 (1995) 75.
35. Ramek, M., Tomic S. and Kojic-Prodic, B., Int. J. Quant. Chem., 60 (1996) 1727.
36. Tomic S., Ramek, M. and Kojic-Prodic, B.,
Combined ab initio SCF and molecular
mechanics studies of propionic
and isobutyric acids and their indole derivatives related
to the phytohormone auxin
(indole-3-acetic acid), accepted for publication in Croat.
Chem. Acta 3 (1998).
37. Maple J. R., Thacher T. S., Dinur U., Hagler A.
T., Chem. Design Autom. News, 5(9)
(1990) 5.
38. BIOSYM (1995) DISCOVER, version 2.97, Biosym Technologies,
10065 Barnes
Canyon Rd., San Diego CA
92121, USA.
39. Kearsley S.K., Smith G.M., Tetrahedron Computer Methodology, 3 (1990) 615.
40. GRID user manual, edition 14, Molecular Discovery
Ltd., West Way House, Elms
Parade, Oxford OX2 9LL,
England
41. Carbo, R., and Calabuig B., Int. J. Quant. Chem., 42 (1992) 1681.
42. Hodgkin E. E. and Richards W. G., Int. J. Quant.
Chem.: Quant. Biol. Symp., 14 (1987)
105.
43. Findlay, S.P. and Dougherty, G. J. Biol. Chem. 1883 (1950) 361.
44. Koegl F.and Kostermans D. G. F. R. Hoppe Seyler's Z. physiol. Chem. 235,(1935) 210.
45. Ramek M. and Tomic S. RHF Conformational Analysis
of the Auxin Phytohormones
n-Ethyl-Indole-3-Acetic
Acid (n=4,5,6), accepted for publication in Int. J. Quant. Chem. .
46. Ramek M., private communication
47. Dauber-Osguthorpe P., Roberts V. A., Osguthorpe
D. J., Wolff J.,
Genest M., Hagler A. T.,
Proteins: Structure, Function and Genetics, 4 (1988) 31.
{kind=link}