SDA
Simulation
of Diffusional Association
(version
6)
Detailed description of input
parameters
# BLOCK 01: dseed
dseed = random number generator seed - defines how the simulations start - different numbers will result in different sets of trajectories. This parameter can be given as the first command line parameter to sda - then the value from the command line will be used.
# BLOCK 02 - BD trajectory parameters: nrun, nprint, nrec, timemax
nrun = number of runs (trajectories). The higher this number the higher is the number of reactive trajectories, nrun_reactive. The relative error of the calculated rate constant is ~ 1/sqrt(nrun_reactive). This means that to have the same relative error, a larger value of nrun is required in cases with lower association rate constants. This parameter can be given as the second command line parameter to sda - then the value from the command line will be used.
nprint = frequency with which rate information is printed: the fraction of reactive trajectories is printed every nprint runs. This is most useful when the simulation takes a long time, then intermediate results are available after each nprint trajectories.
nrec = number of the trajectory to be recorded (nrec > nrun or nrec equals 0 means no recording). If one wants to record the reactive trajectory, it is useful to first run SDA with nprint=1, identify a trajectory in which the number of reactive trajectories changes, and re-run SDA with nrec set to the number of that trajectory. To record all trajectories, set this value to -1. This will use a lot of space.
timemax = maximum length of computed trajectories in ps
# BLOCK 03 - filenames for solute structures : p1f, p2f
p1f = pdb filename for solute 1 (or for the cluster that describes the solid state surface). Standard pdb format is required and atomic radii are assigned according to the character at position 13 or position 14, if the character at position 13 is a space. The atomic radius assignment routine is in van.f - it can be modified to assign different radii. In the case of protein-metal surface docking (ibox>0, imgch=1), the surface atoms should be placed at z=0 (if not solute 1 is automatically shifted so that the atoms with maximum z values are shifted to z=0).
p2f = pdb filename for solute 2
Note: If
Lennard-Jones terms are included in the calculations (apfct != 0), the
pdb file of the solute/surface should also include the probe site identifier given in BLOCK
13 as a number in columns 59-63.
For example, a gold surface can be described by two
kinds of atoms (Av – surface atoms, and Au – bulk atoms) with different
LJ parameters. In this case, two LJ grids are calculated for p2f and the last
column of the rows of the p1f file should give the probe site identifier number:
ATOM 2760 Av CEN
2760 99.090 98.716 0.000 1 001
ATOM 2761 Au CEN
2761 0.640 1.740 -2.400
1 002
Here, Av and Au are two types of surface atom, numbered as the 1st and
2nd probe atoms, respectively.
The probe site information in solute 1 (2) is used only if napt2 (napt1) != 0 (napt1 =0 by default) in Block 13.
# BLOCK 04 - parameters for the description of the solution: probep, probew, hexcl, thres
probep = radius of the probe representing the surface atoms of the solutes. Namely, proteins are represented as a collection of atoms with solvent accessibility larger than thres and each of these atoms is assigned a radius of probep (Å). Typical values of this parameter are 1.4-1.9 Å.
probew = radius of solvent probe used to calculate the accessibilities of solute 2's atoms. A recommended value to use is 1.4 Å. This value is especially important if the hydrophobic desolvation term is used, because the calculated accessibilities are used to calculate the hydrophobic desolvation energy and forces.
hexcl = spacing of the exclusion grid with which a solute is represented. hexcl defines the accuracy with which the shape of the solute is described. Typical values are 0.5-1.0 Å for all-atom models of proteins. It needs to be consistent with the timestep used for BD moves - the spatial accuracy of the protein representation should be comparable to an average BD move in realistic simulations.
thres = accessibility threshold - only the atoms of solutes with accessibility larger than thres (Å) are designated as surface atoms. Moreover, only these atoms are used in calculating hydrophobic interactions.
# BLOCK 05- diffusion parameters for solutes: dm, dr, dr1, irot2f
dm = relative translational diffusion coefficient in Å2/ps. It equals the sum of translational diffusion constants of the two simulated solutes, when both of them are free to move, and equals the translational constant of the mobile solute (2), if the first solute is fixed. For example, if the association of 2 lysozyme molecules in pure water is simulated, then dm should be 0.022 Å2/ps, because the value of the translational diffusion constant for lysozyme in water is 0.011 Å2/ps. According to the Einstein-Stokes formula, the translational diffusion constant scales inversely proportionally to the solvent viscosity (which itself has a complicated dependence on the temperature), and inversely proportionally to the (hydrodynamic) radius of a solute. 1 Å2/ps = 10-8 m2/s = 10-4 cm2/s
dr = rotational diffusion coefficient for solute 2 in radian2/ps. This is inversely proportional to the square of the solute radius. dr is inversely proportional to the solvent viscosity. For lysozyme, for example, dr=2.6*10-5 radian2/ps.
dr1 = rotational diffusion coefficient of solute 1. If irot2f=0i, then this parameter is not used.
irot2f = switch to define whether the first solute rotates. 0 means no rotation and dr1 is not used. irot2f>0 means that the first solute rotates. In all cases, output coordinates are written such that solute 1 does not move and solute 2 moves relativively to solute 1 and its coordinates are recorded.
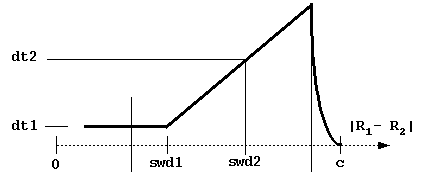
# BLOCK 06 - Brownian motion parameters: dt1, swd1, dt2, swd2, rswd
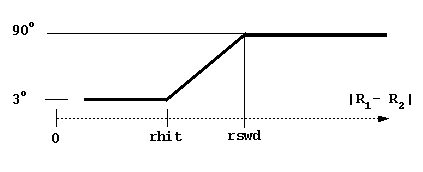
dt1 = basal simulation timestep (in ps, picoseconds). This is the smallest timestep used and is employed when the center-to-center separation is less than swd1 or the distance at which the solutes have the possibility to contact (i.e. the center-to-center distance between solutes is less than rhit = the sum of maximal extents + probe radius + maximal radius of solute 1 atoms).
swd1 = center-to-center distance (Å) at which the timestep starts to increase.
dt2 = timestep at distance swd2.
swd2 = center-to-center distance at which
timestep equals dt2.
These parameters define the variable
timestep designed to accelerate simulations. As shown in the figure below,
the timestep is automatically decreased to zero near the c-surface to reduce
artifacts due to truncation of the trajectories.
Although the profile of the time step influences the computed rates,
the most important parameter is dt1.
Take care not to choose a too small value for this parameter
(since the computing time scales almost linearly with it).
The physical parameter sqrt(dt*dm) measures an average Brownian dynamics step length.
This must be less than 1 Å, which is the characteristic distance describing the roughness
of any protein surface since the atomic bond lengths and van der Waals radii define this scale.
Moreover, dt1 should be less than the characteristic distance over which the interaction forces
vary substantially, which means that the optimal value is influenced by interaction force strength:
one needs smaller time steps for stronger interacting proteins.
rswd = rotation switch distance (see figure below, rhit here is again the distance at which the solutes have the possibility to come into contact). The rotations are accumulated when the time step is small to make the simulations faster. The rotation is carried out only when the accumulated random component of rotation is ~ 3 degrees at small interprotein separations and ~90 degrees at separations larger than rswd.
# BLOCK 07- parameters for dealing with protein-protein separations and close contacts: b, c, rboost, novers, djump
b = start sphere radius (if ibox=0) or distance along z coordinate (ibox >0) (in Å). Simulations are started by placing the center of the second solute at the distance b . For calculation of association constants, there should be no interactions between the solutes at this distance. With finite size grids, b can be selected to be larger than the sum of the solute 1 (or 2) grid extent and the solute 2 (or 1) radius.
c = end sphere radius (if ibox=0) or r distance along z coordinate (ibox > 0) (in Å). Simulation of trajectories is stopped at separation c between the solutes' centers (ibox=0) or when the center of solute 2 has a z coordinate greater than c.
rboost = boost distance (in Å) to move the solutes apart when more than novers moves were not accepted due to overlaps. Boosting is introduced to avoid infinite trajectories and can influence simulation results because trajectories are modified by boosts. The number of boosts is reported in the output log.
novers = number of overlaps allowed before making a boost. Recommended value: 100-200. With a value in this range, boosts are usually not necessary unless the interactions are very strong. This can be the case if a very small probe size (probep) is chosen. Many boosts indicate erroneous input.
djump = threshold for the move length (in Å). Moves in one timestep greater than this distance are counted (not used)
# BLOCK 08- reaction criteria (some parameters are given in BLOCK 09): icommrxn, dind
icommrxn = reaction condition switch. A table with useful settings and meaningful combinations with other parameters is given here.
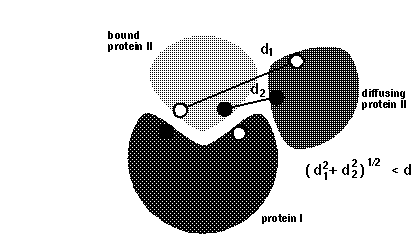
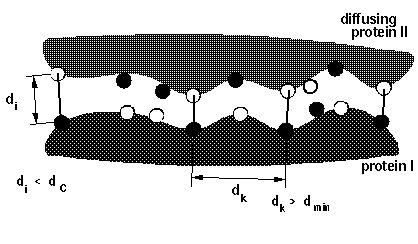
dind = minimal distance between atoms on the same solute defining contacts for satisfying reaction criteria (see d_min in figure below). This parameter is used for icommrxn=3, 6 and and 8. Default value: 6 (Å). For other values of icommrxn, it is equivalent to dind=0.
icommrxn=1 -
RMS average of
all pairwise
distances between pairs of atoms given in file rxna12f.
Writes complexes if d <
windows(nwrec), see BLOCK 09.
icommrxn=2 -
used for association rate calculations.
minimum
pairwise distance between pair of atoms given in files
rxna12f, simultaneous occurrence is monitored for
any 1, 2, 3 or 4 pairs.
Writes complexes if the number of
contacts to define an encounter complex is equal to ncec (see BLOCK 09) AND
windows is equal to win(nwrec). CAN be modifed to less than win(nwrec).
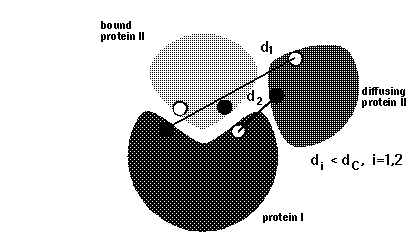
icommrxn=3 -
used for association rate calculations, variant of icommrxn=2.
minimal pairwise distance between a pair of atoms given in file
rxna12f, for independent pairs (defined by dind);
simultaneous occurrence is monitored for any 1, 2, 3 or 4 pairs.
Writes complexes if the number of contacts to define an encounter complex is equal
to ncec (see BLOCK 09) AND windows is equal to win(nwrec). CAN be
modifed to less than win(nwrec).
Better than
icommrxn=2, because it defines an encounter complex more accurately.
>
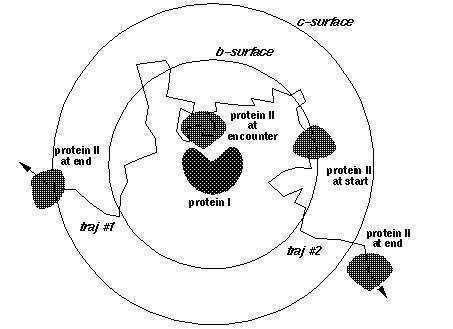
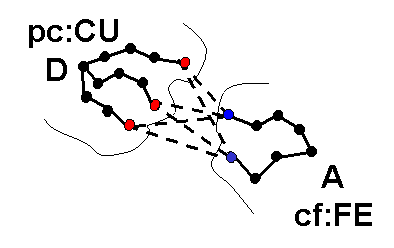
icommrxn=4 - used for electron transfer rate constant calculation. Files rxna1f and rxna2f (defining the reaction atom pairs) should contain pairs of atoms that may take part in an electron transfer path together with their coupling to the donor or acceptor site. In the image below, each possible pairing (broken line) between surface atoms of proteins 1 (blue) and 2 (red) should be defined in files rxna1f and rxna2f.
icommrxn=5 -
used for docking (no association rate calculation).
Accepts all
configurations as
encounter complexes. By using this criterion, the energetically most favorable
complexes will be written to the 'complexes' file (independent of
constraints).
icommrxn=6 -
used for docking (no association rate calculation). Formerly icommrxn=3
in sdac.
Writes complexes after checking all reaction
pairs in *rxna file:
1: all specific pairs must be satisfied
2: at least nnnons (no. of
independent non-specific constraints (depending on dind)) constraints must be
satisfied.
icommrxn=7 -
used for docking (no association rate calculation). Formerly icommrxn=2
in sdac.
Any non-specific constraints are considered, independence is not checked
(equivalent to icommrxn=7 with dind =0, but faster).
icommrxn=9 – used for docking to the surface
(ibox>0, no association rate calculation).
The same as
icommrxn=7, but only the vertical distance (in the z direction) is
checked.
# BLOCK 09- parameters for calculation of protein kinetics (some parameters are included in BLOCK 08): win0, dwin, nwin, iwrec, nwrec, ncec, rxna12f, nnnos, ncmx, rmsd, ionerun, wbmax, ebmin
win0 = reaction distance window minimum value (in Å)
dwin = reaction distance window step (in Å).
nwin = number of reaction distance windows.
iwrec = switch (0 or 1) to specify if complexes satisfying reaction conditions should be recorded. When iwrec=1, the positions of solute 2 will be written to the file "complexes"
nwrec = number of the window up to which the complexes are recorded. For example, in the situation below, nwin=5 reaction distance windows are defined, and rates for forming contacts at each of these 5 distances will be monitored. Encounter complexes with contact distances less than win0+dwin*(nwrec-1), will be recorded if iwrec is set to 1. E.g. in the figure below nwrec=4 is represented. This parameter is only relevant for writing complexes when iwrec=1.
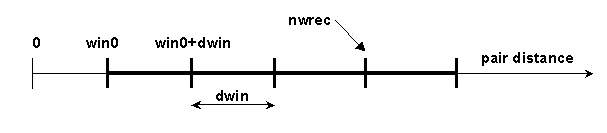
ncec = number of contacts to define an encounter complex; by default set to 2. This parameter is only relevant for writing complexes when iwrec=1 and an association rate calculation (icommrxn=2 or 3) is performed.
rxna12f = filename for reaction atoms. Each
line gives a pair of reaction atoms and the separation between them in Å.
File format:
atom-of-solute-1-from-pdb-file | distance-between-atoms |
atom-of-solute-2-from-pdb-file
A script (convert_sda5_to_sda6_input) converting the two *.rxna files used in the SDA 5 program to
the new format used in SDA6 can be found in the
sda/auxi directory. Reaction atoms do
not have to be real atoms, although in this file these atoms/points
should be given like atoms in PDB format. Different combinations
of the same atoms/points can be prepared by editing these reaction atom
files.
nnnons = no. of nonspecific constraints to satisfy.
ncmx = no. of distinct lowest energy configurations to write (to 'complexes')
rmsd = minimum rmsd between complexes in Å. During the BD simulations, if a new docking pose is considered similar to a previously stored pose, then the configuration with the lowest total energy is stored and the counter for this docking pose is increased. A pose is considered similar to a previous pose if they have an RMSD less than the designated threshold (parameter rmsd).
ionerun = if rmsd check is to be done for one run (1) or for all
--------------------
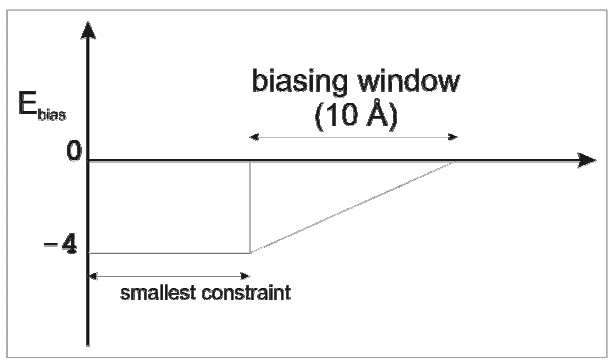
Further parameters to specify for biased BD:
wbmax = pair distance increment to begin biased BD.
ebmin = biasing energy. The biasing parameter, Ebias, changes linearly from ebmin at the beginning of the window.
Biasing (based on a Metropolis algorithm) can be used to favor the BD steps that lead towards the fulfillment of distance constraints. Biasing is applied within a “biasing window” from a distance equal to the smallest distance constraint plus 10 Å. The biasing parameter, Ebias, changes linearly from ebmin (=0) at the beginning of the window to -4 at the smallest distance constraint. Therefore, if ΔEbias (Ebias at the new BD step - Ebias at the previous BD step) ≤ 0, that is if the proteins move towards fulfillment of the distance constraint, the new step is always accepted. Otherwise, the next step is accepted if r < e-βΔEbias, where r is a random number in the interval [0:1] and β = 1 / kT. Note: wbmin is equal to 0 and kT is not considered.
# BLOCK 10 - parameters for electrostatic energy/forces: epfct,
ep1f, ep2f, qef1f, qef2f
epfct = factor by which the electrostatic potential grid values read in are to be multiplied. Should be 1 if no modifications are needed.
ep1f = filename for the electrostatic potential of solute 1 in UHBD binary format. It is assumed that the potentials are written in kcal/mol/e units. Note that APBS generated grids written in UHBD format are in units of kT/e and ascii and as such cannot be used directly with SDA. They should be rescaled and converted with the auxiliary program apbs2uhbd, to bring them into consistency with UHBD.
ep2f = filename for the electrostatic potential of solute 2 in UHBD format, binary.
qef1f = filename for effective charges of solute 1. This is the output of the ECM program. The effective charge representation in variant R (extension _R) is to be used, see detailed description here.
qef2f = filename for effective charges of solute 2
# BLOCK 11 -parameters for calculation electrostatic desolvation energy/forces: edfct, ed1f, ed2f
edfct = factor by which the electrostatic desolvation potential grid values are to be multiplied. The value 1.00 corresponds to the uncorrected electrostatic desolvation penalty for a charge in a solvent due to the low dielectric cavity of a solute treated as a collection of van der Waals spheres. More appropriate factors can be derived by comparing SDA calculated energies of recorded complexes with energies calculated with more accurate methods (solving the finite difference Poisson-Boltzmann equation, for example). Recommended value:1.67 (with solute interior dielectric constant set to 4 and the dielectric boundary between the solute interior and high dielectric solvent defined by the van der Waals surface of the solute). (Gabdoulline, R. R.; Wade, R. C., J. Mol. Biol. 1999, 291, 149-162 )
ed1f = filename for the electrostatic desolvation potential of solute 1 in UHBD format. This grid can be calculated with program mk_ed_grd which is included in the SDA distribution (units in kcal/mol/e).
ed2f = filename for the electrostatic desolvation potential of solute 2 in UHBD format.
# BLOCK 12 -parameters for calculation of hydrophobic desolvation energy/forces: hdfct, hd1f, hd2f
hdfct = factor by which the hydrophobic desolvation potential grid values are to be multiplied. In the SDA program, hydrophobic desolvation energies are calculated by multiplying the solvent accessibilities of the atoms of one of the solutes by the value of the hydrophobic desolvation potential grid calculated for the other solute. If the grid is calculated so that the sum of contributions from all accessible atoms gives the buried solvent accessible area, then realistic values for this factor are in the range -0.0050 to -0.060 kcal/mole/Å2 (or -5 to -60 cal/mole/Å2). The value of this factor can also be derived by comparing SDA calculated energies of recorded complexes with energies calculated with more accurate methods. E.g. values of -0.013 to -0.019 kcal/mole/Å2 were used in Gabdoulline R.R., Wade. R.C. JACS, 2009, 131, 9320
hd1f = filename for the hydrophobic desolvation potential of solute 1 in UHBD format. This grid can be calculated with program mk_hd_grd which is included in SDA distribution.
hd2f = filename for the hydrophobic desolvation potential of solute 2 in UHBD format.
# BLOCK 13 - parameters for calculation of Lennard-Jones energy/forces: apfct, napt1,napt2, ap1f, ap2f,ap2fg1,ap2fg2,... ap2fgx, where x=napt2
apfct = factor by which the LJ grid values are to be multiplied. Should be 1.0 if no modifications are needed.
napt1 = number of LJ grids for solute/surface 1. Currently the LJ grid is calculated only for solute 2 for each probe site (atom type) of solute 1. Thus, napt1 should be zero (default: napt1=0) .
napt2 = number of LJ grids for solute 2 (default napt2=0)
ap2gf1 = filename for the LJ grid potential
of solute 2 for the 1st probe atom of solute/surface 1 in UHBD format.
This grid can be calculated with program mk_LJ_grd which is included in
the SDA distribution here. or, more
easily, with the python script prepLJgrids.py here.
If there are several atom types (probe sites) i.e. napt2>1, all grid
names should be given as ap2gf2, ap2gf3 etc
# BLOCK 14 - dimension of the simulation box for protein-surface docking, ibox>0: xmin, xmax, ymin, ymax, zmin )
xmin = minimum x coordinate of the simulation box (applied to the position of the center of solute 2)
xmax = maximum x coordinate
ymin = minimum y coordinate
ymax = maximum y coordinate
zmin = minimum z coordinate (from the surface of the solid state cluster)
Important: to compute the LJ energy between solute 2 and the solid state surface (or solute 1) properly, the simulation box area in the (x,y) plane should be smaller than the size of the surface (or solute 1) in the (x,y) plane by the radius of solute 1 plus ~10 Å. Otherwise, the simulation box size in the (x,y) plane will be reduced automatically. In the case of protein-metal surface docking (ibox>0, imgch=1 ), surface atoms should be placed at z=0 (otherwise solute 1 will be shifted automatically so that the atoms with maximum z are shifted to z=0).
# BLOCK 15 - parameters of the hydration shell on a solid state surface (currently optimized for a Au(111) surface): zadw, eadw, radw, aadw, ep_scale, ep_scaleA, ep_scaleB, ep_scaleC (see Kokh et al, JCTC, 2010)
zadw = Zadw in Å ; parameter for calculation of the surface desolvation potential Φ
eadw = Φ° in kT/Ų; parameter for calculation of the surface desolvation potential Φ
aadw = α in Å; parameter for calculation of the surface desolvation potential Φ
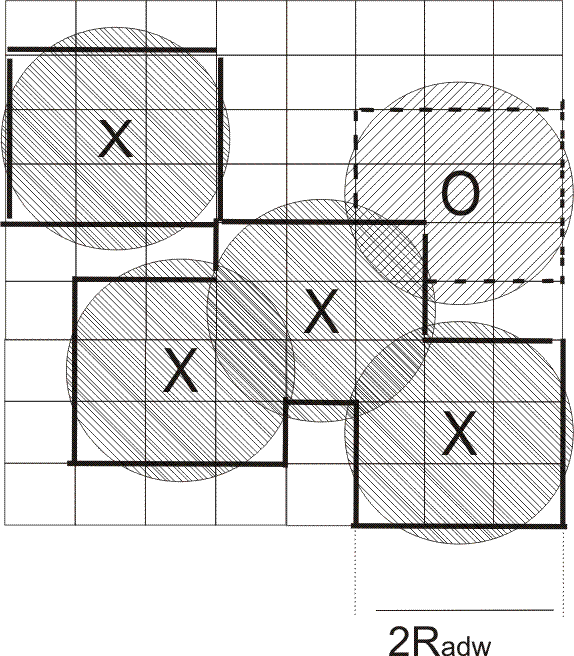
radw = Radw in Å; The effective desolvation radius for defining the desolvated surface area. The surface desolvation energy is calculated as :
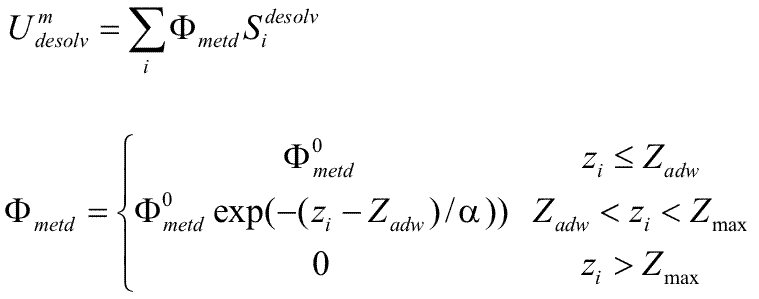
The computed desolvated area is shown by bold lines, the solid and
dashed lines corresponding to water desorption from the first and
second hydration layers, respectively. The circles with crosses
represent close solute atoms and that with a zero is at an intermediate
distance.
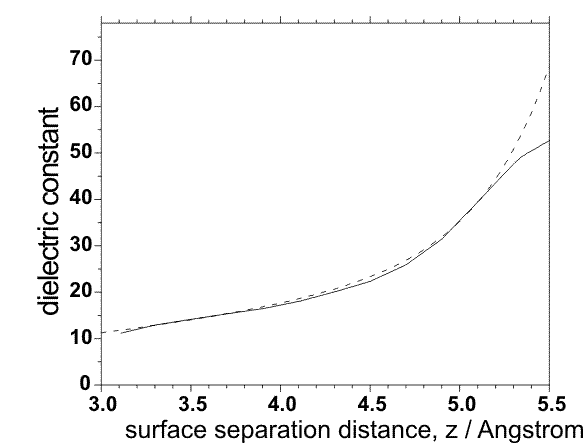
ep_scale = switch (0/1) for scaling of the dielectric constant for the case of the overlapping cavities. Can be used only for protein-surface simulations (ibox=1).
ep_scaleA = A ; Parameter for calculation of scaled dielectric constant; default value: A=0.8 1/Ų
ep_scaleB = B ; default value: B=0.39 1/Ų
ep_scaleC = C ; default value: C=10.4. The dielectric constant is calculated as: ε=4.0+A*z*z+exp(-z/B - C) for z < 5.5 Å
# BLOCK 16- general parameters describing the symmetry of the simulation volume and the type of electrostatic calculation procedure: ibox, imgch, iefast
ibox = switch(0,1,2): ibox=0 - BD simulation in a spherical simulation volume (default); ibox=1 - simulation volume is a box defined by (xmin;xmax), (ymin;ymax), (zmin,b) and is used for simulation of protein association to a solid state surface (note, that at ibox=1 the box size is checked and automatically reduced if the protein is too large for the surface size, see xmin,xmax,ymin,ymax parameters); ibox=2 – periodic simulation box with a period defined by (xmin;xmax), (ymin;ymax).
imgch = image-charge switch. imgch=1 : Instead of Coulomb interaction, the image-charge electrostatic energy is calculated. Used only with ibox=1 for metal surfaces. Default: imgch=0
iefast = electrostatic force switch. If iefast=-1, the fast eforce routine for electrostatic force calculations is used. Default: iefast=0, (eforce routine is not used). The fast routine is only used when there is no desolvation term and no rotation of solute 1. In the fast eforce routine, only forces acting on the second solute from the electrostatic field of the first solute are computed (otherwise the forces equal an average of the interactions of solute 2 on solute 1 and solute 1 on solute 2).
# BLOCK 17 -parameters for PMF calculations (for simulation of protein docking to the solid surface only): free_en, free_en_z, free_en_h
free_en = switch for calculation of the energy landscape, PMF, and adsorption free energy. Can only be used for protein-surface association (ibox=1). Default: free_en=0 - no energy landscape computed. The maximal dimension of the energy landscape is defined in the file maxdim.inc.
For example, for free_en=1
and
parameter(nthmx=90,nfimx=180,nommx=90,ndxmx=6) defined in the
maxdim.inc file,
the grid spacing for protein rotation is (1°,1°,1°) and for translation
in the xy plane is (1 Å,1 Å). If free_en=2, the grid spacing
will be twice as large: (2°,2°,2°,2 Å,2 Å)
The general rule for determining the grid spacing is :
(free_en*90°/nthmx; free_en*180°/nfimx; free_en*90°/nommx) and
(free_en*6Å /ndxmx, free_en*6Å /ndxmx) for rotation and translation,
respectively.
Starting from the distance swd1 (distance between the centers of the protein and the solid state cluster) rotational and translational spacings are increased by 2 and 10 times, respectively.
free_en_z = starting protein-surface distance in Å for computing the energy landscape.
free_en_h = step interval taken in the z direction, in Å, for computing the energy landscape
#BLOCK 18: monitoring of intermediate results fnintr
fnintr = name of the file where intermediate results (fraction of reactive trajectories for every reaction window distance and for forming 1, 2, 3 or 4 contacts at these distances) are written every nprint runs.
Old
format (used in SDA v5.01 and 4.23):
The input file consists of a
series of blocks of input parameters. The format of each block of the
input file is as follows:
A one (!) line description of a block of
input parameters, starting with "#". On the next line,
comma or space separated numerical values are given in the required
order. Then, the file names (if any) are given in the required order,
each on separate lines. An example file looks like this (follow
the
links for detailed descriptions):
# dseed
= random_number_generator_seed
256.d0
# nrun,
nprint, nrec = no_of_runs, printout_every_nprint_run,
record_this_run (or all runs in case of a negative nunber)
100, 5,
0
# p1f, p2f =
pdb_filenames_for_solute/protein_1, 2
p1.pdb
p2.pdb
#
probe, probew, hexcl, thres - probe_pr2(Å),
probe_water(Å), excl_grid_spacing(Å), acc_thres(Å2)
1.70,
1.4, 0.5, 0.0
# dm, dr, dr1, irot2f =
tr_diffco, rot_diffco_2, rot_diffco_1, if_prot_1_rotates
0.027,
3.92e-5, 3.92e-5, 1
# dt1, swd1, dt2, swd2,
rswd = tstep, tstep_sw_dist_1, tstep_2, tstep_sw_dist_2,
rot_sw_dist
0.50, 50.0, 20.0, 90.0, 90.0
# b,
c, rboost, novers, djump = b, c, boost_val, no_overs_bef_boost,
jump_thr_to_rec
100.0, 500.0, 1.0, 150, 5.0
#
icommrxn, dind = reaction_mon_switch,
force_switch, indep_dist
3, 6.0
# win0,
dwin, nwin, iwrec, nwrec, ncec, rxna12f = rxn_defs,
rxn_at_files_prot_1, 2
1.0, 1.0, 50, 1, 40, 500
p12.rxna
#
epfct, ep1f, ep2f, qef1f, qef2f = e-pot_factor,
e-pot_for_prot_1, 2, charges_for_prot_1,
2
1.00
ep1.grd
ep2.grd
p1.echa
p2.echa
#
edfct, ed1f, ed2f = e-deso_factor,
e-deso_file_for_prot_1, 2
1.00
ed1.grd
ed2.grd
#
hdfct, hd1f, hd2f = h-deso_factor, skin,
h-deso_file_for_prot_1,2
1.00
hd1.grd
hd2.grd
#
fnintr = intermediate results
filename
sda.int
$LastChangedDate: 2010-10-06 15:40:58 +0200 (Wed, 06 Oct 2010) $ by $Author: richtesn $